diff --git a/.github/CONTRIBUTING.md b/.github/CONTRIBUTING.md
new file mode 100644
index 0000000..fe79124
--- /dev/null
+++ b/.github/CONTRIBUTING.md
@@ -0,0 +1,262 @@
+# Contributing to CHARLIE
+
+## Proposing changes with issues
+
+If you want to make a change, it's a good idea to first
+[open an issue](https://code-review.tidyverse.org/issues/)
+and make sure someone from the team agrees that it’s needed.
+
+If you've decided to work on an issue,
+[assign yourself to the issue](https://docs.github.com/en/issues/tracking-your-work-with-issues/assigning-issues-and-pull-requests-to-other-github-users#assigning-an-individual-issue-or-pull-request)
+so others will know you're working on it.
+
+## Pull request process
+
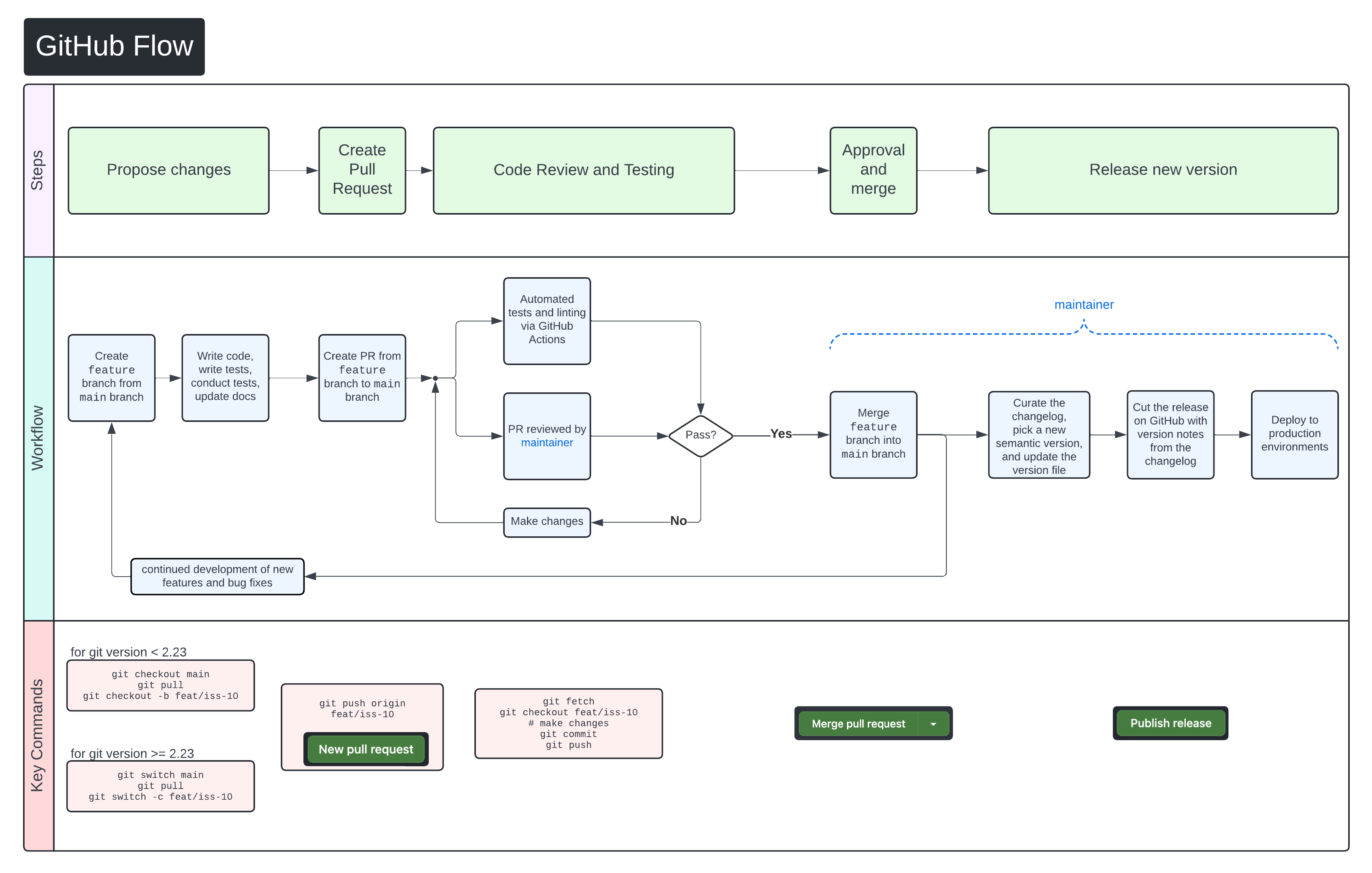
+We use [GitHub Flow](https://docs.github.com/en/get-started/using-github/github-flow)
+as our collaboration process.
+Follow the steps below for detailed instructions on contributing changes to
+CHARLIE.
+
+
+
+### Clone the repo
+
+If you are a member of [CCBR](https://github.com/CCBR),
+you can clone this repository to your computer or development environment.
+Otherwise, you will first need to
+[fork](https://docs.github.com/en/pull-requests/collaborating-with-pull-requests/working-with-forks/fork-a-repo)
+the repo and clone your fork. You only need to do this step once.
+
+```sh
+git clone https://github.com/CCBR/CHARLIE
+```
+
+> Cloning into 'CHARLIE'...
+> remote: Enumerating objects: 1136, done.
+> remote: Counting objects: 100% (463/463), done.
+> remote: Compressing objects: 100% (357/357), done.
+> remote: Total 1136 (delta 149), reused 332 (delta 103), pack-reused 673
+> Receiving objects: 100% (1136/1136), 11.01 MiB | 9.76 MiB/s, done.
+> Resolving deltas: 100% (530/530), done.
+
+```sh
+cd CHARLIE
+```
+
+### If this is your first time cloning the repo, you may need to install dependencies
+
+- Install snakemake and singularity or docker if needed (biowulf already has these available as modules).
+
+- Install the python dependencies with pip
+
+ ```sh
+ pip install .
+ ```
+
+ If you're developing on biowulf, you can use our shared conda environment which already has these dependencies installed
+
+ ```sh
+ . "/data/CCBR_Pipeliner/db/PipeDB/Conda/etc/profile.d/conda.sh"
+ conda activate py311
+ ```
+
+- Install [`pre-commit`](https://pre-commit.com/#install) if you don't already
+ have it. Then from the repo's root directory, run
+
+ ```sh
+ pre-commit install
+ ```
+
+ This will install the repo's pre-commit hooks.
+ You'll only need to do this step the first time you clone the repo.
+
+### Create a branch
+
+Create a Git branch for your pull request (PR). Give the branch a descriptive
+name for the changes you will make, such as `iss-10` if it is for a specific
+issue.
+
+```sh
+# create a new branch and switch to it
+git branch iss-10
+git switch iss-10
+```
+
+> Switched to a new branch 'iss-10'
+
+### Make your changes
+
+Edit the code, write and run tests, and update the documentation as needed.
+
+#### test
+
+Changes to the **python package** code will also need unit tests to demonstrate
+that the changes work as intended.
+We write unit tests with pytest and store them in the `tests/` subdirectory.
+Run the tests with `python -m pytest`.
+
+If you change the **workflow**, please run the workflow with the test profile
+and make sure your new feature or bug fix works as intended.
+
+#### document
+
+If you have added a new feature or changed the API of an existing feature,
+you will likely need to update the documentation in `docs/`.
+
+### Commit and push your changes
+
+If you're not sure how often you should commit or what your commits should
+consist of, we recommend following the "atomic commits" principle where each
+commit contains one new feature, fix, or task.
+Learn more about atomic commits here:
+
+
+First, add the files that you changed to the staging area:
+
+```sh
+git add path/to/changed/files/
+```
+
+Then make the commit.
+Your commit message should follow the
+[Conventional Commits](https://www.conventionalcommits.org/en/v1.0.0/)
+specification.
+Briefly, each commit should start with one of the approved types such as
+`feat`, `fix`, `docs`, etc. followed by a description of the commit.
+Take a look at the [Conventional Commits specification](https://www.conventionalcommits.org/en/v1.0.0/#summary)
+for more detailed information about how to write commit messages.
+
+```sh
+git commit -m 'feat: create function for awesome feature'
+```
+
+pre-commit will enforce that your commit message and the code changes are
+styled correctly and will attempt to make corrections if needed.
+
+> Check for added large files..............................................Passed
+> Fix End of Files.........................................................Passed
+> Trim Trailing Whitespace.................................................Failed
+>
+> - hook id: trailing-whitespace
+> - exit code: 1
+> - files were modified by this hook
>
+> Fixing path/to/changed/files/file.txt
>
+> codespell................................................................Passed
+> style-files..........................................(no files to check)Skipped
+> readme-rmd-rendered..................................(no files to check)Skipped
+> use-tidy-description.................................(no files to check)Skipped
+
+In the example above, one of the hooks modified a file in the proposed commit,
+so the pre-commit check failed. You can run `git diff` to see the changes that
+pre-commit made and `git status` to see which files were modified. To proceed
+with the commit, re-add the modified file(s) and re-run the commit command:
+
+```sh
+git add path/to/changed/files/file.txt
+git commit -m 'feat: create function for awesome feature'
+```
+
+This time, all the hooks either passed or were skipped
+(e.g. hooks that only run on R code will not run if no R files were
+committed).
+When the pre-commit check is successful, the usual commit success message
+will appear after the pre-commit messages showing that the commit was created.
+
+> Check for added large files..............................................Passed
+> Fix End of Files.........................................................Passed
+> Trim Trailing Whitespace.................................................Passed
+> codespell................................................................Passed
+> style-files..........................................(no files to check)Skipped
+> readme-rmd-rendered..................................(no files to check)Skipped
+> use-tidy-description.................................(no files to check)Skipped
+> Conventional Commit......................................................Passed
> [iss-10 9ff256e] feat: create function for awesome feature
+> 1 file changed, 22 insertions(+), 3 deletions(-)
+
+Finally, push your changes to GitHub:
+
+```sh
+git push
+```
+
+If this is the first time you are pushing this branch, you may have to
+explicitly set the upstream branch:
+
+```sh
+git push --set-upstream origin iss-10
+```
+
+> Enumerating objects: 7, done.
+> Counting objects: 100% (7/7), done.
+> Delta compression using up to 10 threads
+> Compressing objects: 100% (4/4), done.
+> Writing objects: 100% (4/4), 648 bytes | 648.00 KiB/s, done.
+> Total 4 (delta 3), reused 0 (delta 0), pack-reused 0
+> remote: Resolving deltas: 100% (3/3), completed with 3 local objects.
+> remote:
+> remote: Create a pull request for 'iss-10' on GitHub by visiting:
+> remote: https://github.com/CCBR/CHARLIE/pull/new/iss-10
+> remote:
+> To https://github.com/CCBR/CHARLIE
>
> [new branch] iss-10 -> iss-10
+> branch 'iss-10' set up to track 'origin/iss-10'.
+
+We recommend pushing your commits often so they will be backed up on GitHub.
+You can view the files in your branch on GitHub at
+`https://github.com/CCBR/CHARLIE/tree/`
+(replace `` with the actual name of your branch).
+
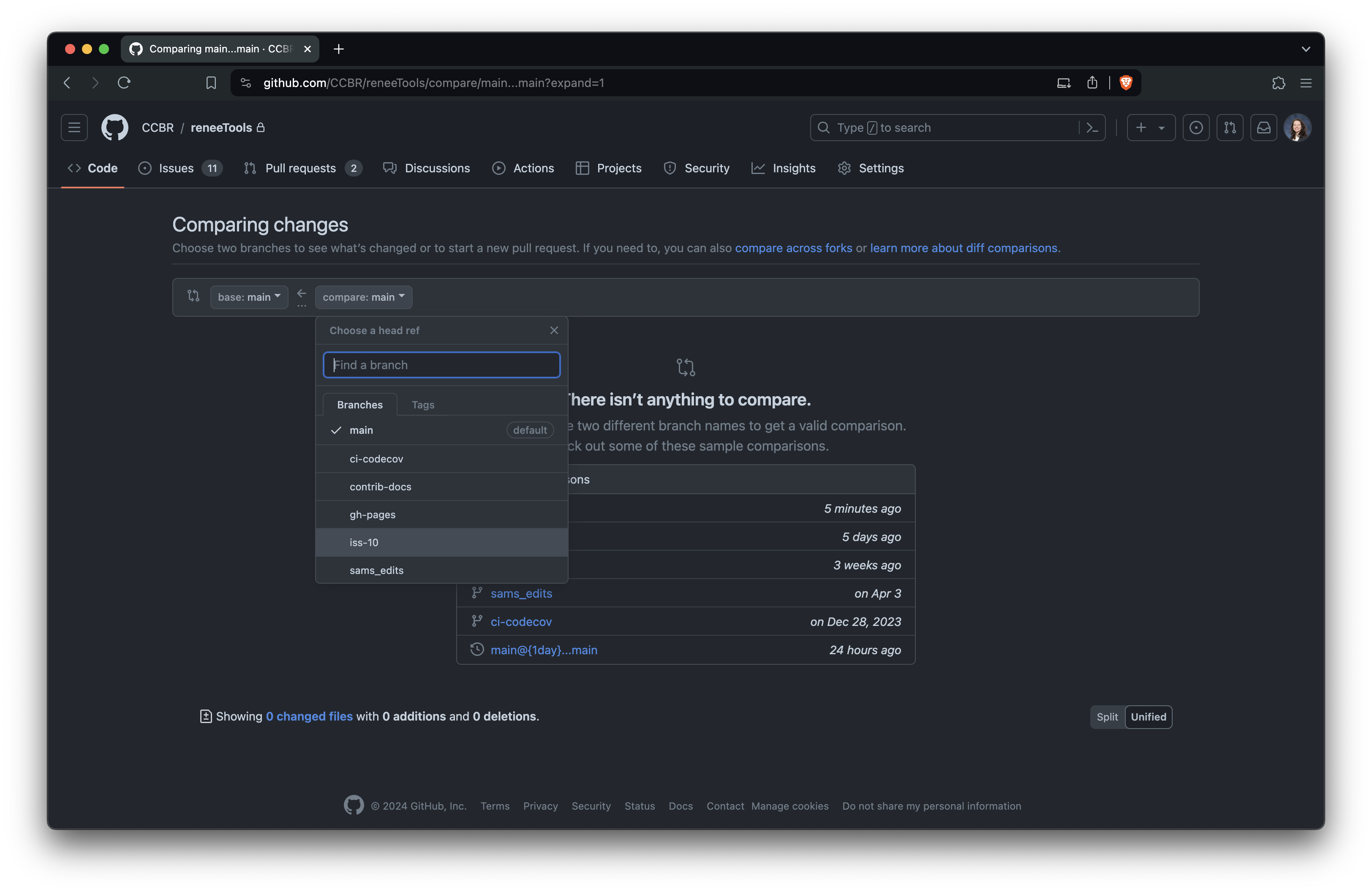
+### Create the PR
+
+Once your branch is ready, create a PR on GitHub:
+
+
+Select the branch you just pushed:
+
+
+
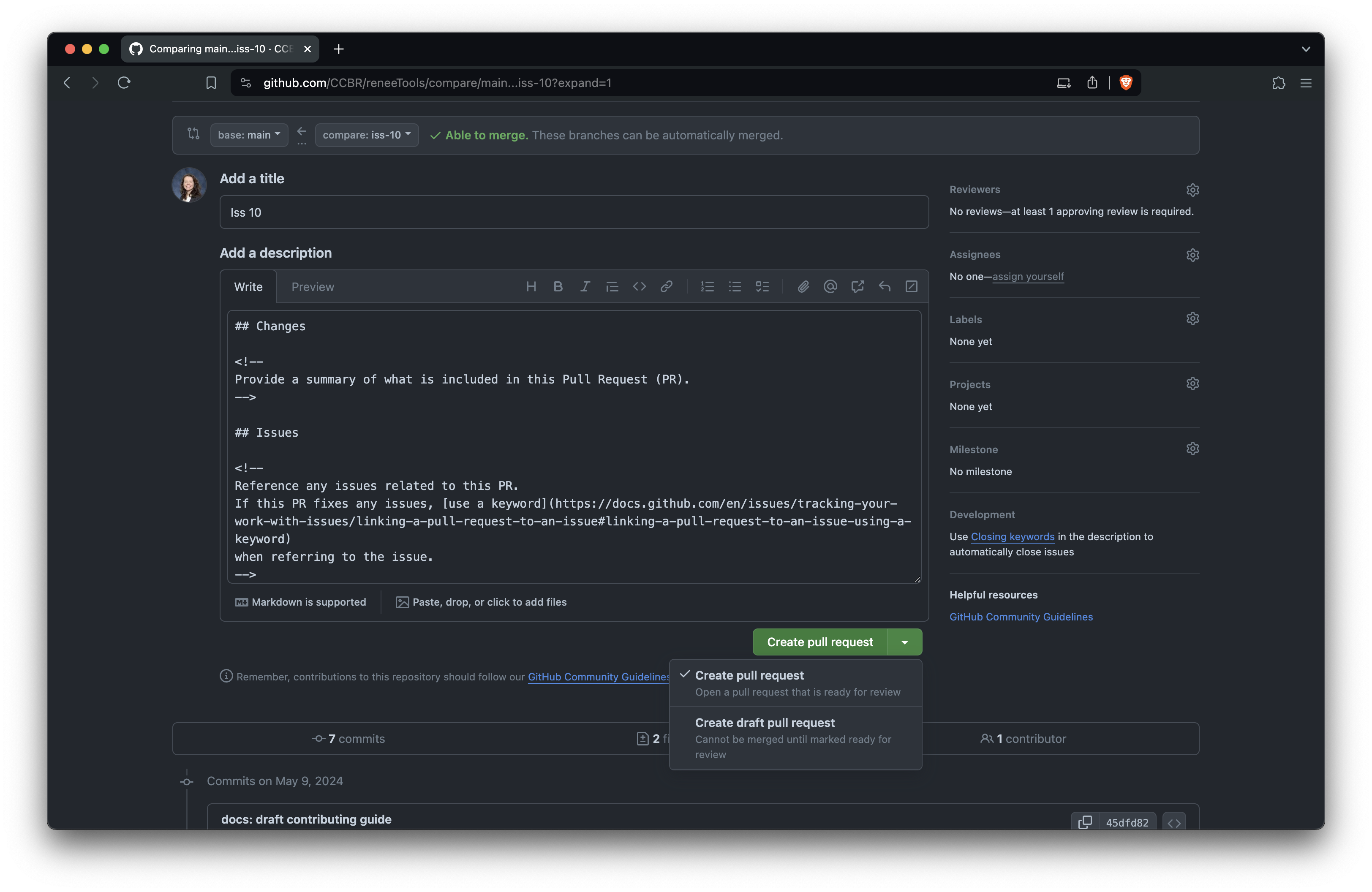
+Edit the PR title and description.
+The title should briefly describe the change.
+Follow the comments in the template to fill out the body of the PR, and
+you can delete the comments (everything between ``) as you go.
+Be sure to fill out the checklist, checking off items as you complete them or
+striking through any irrelevant items.
+When you're ready, click 'Create pull request' to open it.
+
+
+
+Optionally, you can mark the PR as a draft if you're not yet ready for it to
+be reviewed, then change it later when you're ready.
+
+### Wait for a maintainer to review your PR
+
+We will do our best to follow the tidyverse code review principles:
+.
+The reviewer may suggest that you make changes before accepting your PR in
+order to improve the code quality or style.
+If that's the case, continue to make changes in your branch and push them to
+GitHub, and they will appear in the PR.
+
+Once the PR is approved, the maintainer will merge it and the issue(s) the PR
+links will close automatically.
+Congratulations and thank you for your contribution!
+
+### After your PR has been merged
+
+After your PR has been merged, update your local clone of the repo by
+switching to the main branch and pulling the latest changes:
+
+```sh
+git checkout main
+git pull
+```
+
+It's a good idea to run `git pull` before creating a new branch so it will
+start from the most recent commits in main.
+
+## Helpful links for more information
+
+- [GitHub Flow](https://docs.github.com/en/get-started/using-github/github-flow)
+- [semantic versioning guidelines](https://semver.org/)
+- [changelog guidelines](https://keepachangelog.com/en/1.1.0/)
+- [tidyverse code review principles](https://code-review.tidyverse.org)
+- [reproducible examples](https://www.tidyverse.org/help/#reprex)
+- [nf-core extensions for VS Code](https://marketplace.visualstudio.com/items?itemName=nf-core.nf-core-extensionpack)
diff --git a/.github/workflows/docs-mkdocs.yml b/.github/workflows/docs-mkdocs.yml
new file mode 100644
index 0000000..89b719b
--- /dev/null
+++ b/.github/workflows/docs-mkdocs.yml
@@ -0,0 +1,25 @@
+name: docs
+on:
+ workflow_dispatch:
+ release:
+ types:
+ - published
+ push:
+ branches:
+ - main
+ paths:
+ - "docs/**"
+ - "**.md"
+ - .github/workflows/docs-mkdocs.yml
+ - mkdocs.yml
+
+jobs:
+ mkdocs:
+ runs-on: ubuntu-latest
+ steps:
+ - uses: actions/checkout@v4
+ with:
+ fetch-depth: 0
+ - uses: CCBR/actions/mkdocs-mike@v0.1
+ with:
+ github-token: ${{ github.token }}
diff --git a/.github/workflows/projects.yml b/.github/workflows/projects.yml
deleted file mode 100644
index 61a2816..0000000
--- a/.github/workflows/projects.yml
+++ /dev/null
@@ -1,14 +0,0 @@
-name: Add issues/PRs to user projects
-
-on:
- issues:
- types:
- - assigned
- pull_request:
- types:
- - assigned
-
-jobs:
- add-to-project:
- uses: CCBR/.github/.github/workflows/auto-add-user-project.yml@v0.1.0
- secrets: inherit
diff --git a/CHANGELOG.md b/CHANGELOG.md
index dc577a5..e5bc67e 100644
--- a/CHANGELOG.md
+++ b/CHANGELOG.md
@@ -1,19 +1,20 @@
-# CHARLIE development version
+## CHARLIE development version
- Major updates to convert CHARLIE from a biowulf-specific to a platform-agnostic pipeline (#102, @kelly-sovacool):
- All rules now use containers instead of envmodules.
- Default config and cluster config files are provided for use on biowulf and FRCE.
- New entry `TEMPDIR` in the config file sets the temporary directory location for rules that require transient storage.
- New `--singcache` argument to provide a singularity cache dir location. The singularity cache dir is automatically set inside `/data/$USER/` or `$WORKDIR/` if `--singcache` is not provided.
+- Minor documentation improvements. (#114, @kelly-sovacool)
-# CHARLIE 0.10.1
+## CHARLIE 0.10.1
- strand are reported together, strand from all callers are reported,
- both + and - flanking sites are reported,
- rev-comp function updated,
- updated versions of tools to match available tools on BIOWULF.
-# CHARLIE 0.9.0
+## CHARLIE 0.9.0
Significant upgrades since the last release:
@@ -34,35 +35,35 @@ Significant upgrades since the last release:
- multitude of comments throughout the snakefiles including listing of output file column descriptions
- preliminary GH actions added
-# CHARLIE 0.7.0
+## CHARLIE 0.7.0
- 5 circRNA callers
- all-sample counts matrix with annotations
-# CHARLIE 0.6.9
+## CHARLIE 0.6.9
- Optimized pysam scripts
- fixed premature completion of singularity rules
-# CHARLIE 0.6.5
+## CHARLIE 0.6.5
- updated config.yaml to use the latest HSV-1 annotations received from Sarah (050421)
-# CHARLIE 0.6.4
+## CHARLIE 0.6.4
- create linear reads BAM file
- create linear reads BigWigs for each region in the .regions file.
-# CHARLIE 0.6.3
+## CHARLIE 0.6.3
- QOS not working for Taka... removed from cluster.json
- recall rule requires python/3.7 ... env module updated
-# CHARLIE 0.6.2
+## CHARLIE 0.6.2
- BSJ files are in BSJ subfolder... bug fix for v0.6.1
-# CHARLIE 0.6.1
+## CHARLIE 0.6.1
- customBSJs recalled from STAR alignments
- only for PE
@@ -70,6 +71,44 @@ Significant upgrades since the last release:
- create sense and anti-sense BSJ BAMs and BW for each reference (host+viruses)
- find reads which contribute to CIRI BSJs but not on the STAR list of BSJ reads, see if they contribute to novel (not called by STAR) BSJs and append novel BSJs to customBSJ list
-# CHARLIE 0.6.0
+## CHARLIE 0.6.0
cutadapt_min_length to cutadapt rule... setting it to 15 in config (for miRNAs, Biot and short viral features)
+
+## CHARLIE 0.5.0
+
+- `run_clear` is now set to True (as default)
+- `circ_quant` replaces `clear_quant` in the CLEAR rule. In order words, we are reusing the STAR alignment file and the circExplorer2 output file for running CLEAR. No need to run HISAT2 and TopHat (fusion-search with Bowtie1). This is much quicker.
+- Using picard to estimate duplicates using[ _MarkDuplicates_](https://gatk.broadinstitute.org/hc/en-us/articles/360037052812-MarkDuplicates-Picard-)
+- Generating a per-run [multiqc](https://multiqc.info/) HTML report
+- Using [_eulerr_](https://www.rdocumentation.org/packages/eulerr/versions/6.1.0) R package to generate CIRI-CircExplorer circRNA Venn diagrams and include them in the mulitqc report
+- Gather per job cluster metadata like queue time, run time, job state etc. Stats are compiled in **HPC_summary** file
+- CLEAR pipeline _quant.txt_ file is annotated for known circRNAs
+- `WORKDIR` can now be a relative path
+- bam2bw conversion fix for BSJ and spliced_reads. [Issue](https://github.com/kopardev/circRNA/issues/17) closed!
+
+## CHARLIE 0.4.0
+
+- [CLEAR](https://github.com/YangLab/CLEAR) added.
+- wrapper script (`run_circrna_daq.sh`) added for local and cluster execution.
+- "spliced reads only" bam created and split by regions
+
+## CHARLIE 0.3.0
+
+- Lookup table for hg38 to hg19 circRNA annotations is updated... this eliminate one-to-many hits from the previous version
+- BSJs extracted as different bam file.
+- flowchart added
+- adding slurmjobid to log/err file names
+- v0.3.1 has significant (>10X) performance improvements at BSJ bam creation
+- v0.3.3 splits BSJ bams into human and viral bams, and also converts them to bigwigs
+- v0.3.4 adds hg38_rRNA_masked_plus_rRNA_plus_viruses_plus_ERCC reference (source:Sarah)
+
+## CHARLIE 0.2.0
+
+- SE support added .. PE/SE samples handled concurrently
+- `envmodules` used in Snakemake in place of `module load` statements
+
+## CHARLIE 0.1.0
+
+- base version
+- PE only support
diff --git a/README.md b/README.md
index 048286c..ed0f3c5 100644
--- a/README.md
+++ b/README.md
@@ -1,10 +1,17 @@
# CHARLIE
-
+**C**ircrnas in **H**ost **A**nd vi**R**uses ana**L**ysis p**I**p**E**line
+
+[](https://github.com/CCBR/CHARLIE/actions/workflows/build.yml)
+[](https://github.com/CCBR/CHARLIE/issues)
+[](https://github.com/CCBR/CHARLIE/pulls)
+
+
+[](https://github.com/CCBR/CHARLIE/blob/main/LICENSE)
### Table of Contents
-- [CHARLIE - **C**ircrnas in **H**ost **A**nd vi**R**uses ana**L**ysis p**I**p**E**line](#charlie)
+- [CHARLIE](#charlie)
- [Table of Contents](#table-of-contents)
- [1. Introduction](#1-introduction)
- [2. Flowchart](#2-flowchart)
@@ -45,11 +52,11 @@ This circularRNA detection pipeline uses CIRCExplorer2, CIRI2 and many other too
### 2. Flowchart
-
+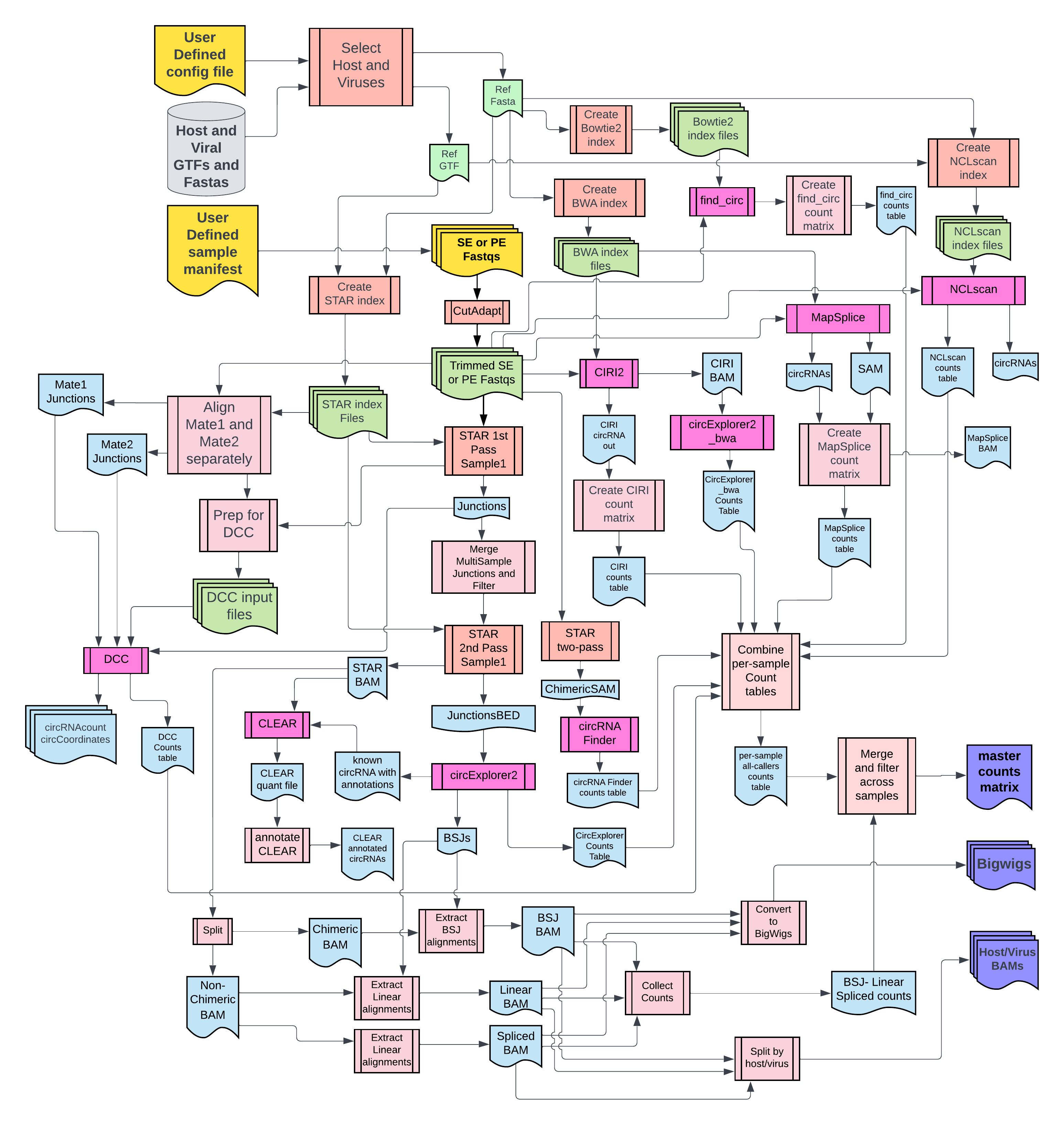
-For complete documentation with tutorial go [here](https://CCBR.github.io/CHARLIE/).
+For complete documentation, view the website .
-> DISCLAIMER: New circRNA tools have been added CHARLIE and the documentation is currently out of date!
+> ⚠️ DISCLAIMER: New circRNA tools have been added CHARLIE and the documentation is currently out of date!
### 3. Software Dependencies
@@ -165,9 +172,9 @@ Optional Arguments:
Example commands:
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/ouput/folder -m=init
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/ouput/folder -m=dryrun
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/ouput/folder -m=run
+ bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=init
+ bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=dryrun
+ bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=run
##########################################################################################
@@ -236,7 +243,7 @@ This will create the reference fasta and gtf file based on the selections made i
#### Run
-If `-m=dryrun` was sucessful, then simply do `-m=run`. The output will look something like this
+If `-m=dryrun` was successful, then simply do `-m=run`. The output will look something like this
```
... ... skipping ~1000 lines
diff --git a/docs/CHANGELOG.md b/docs/CHANGELOG.md
new file mode 100644
index 0000000..bebc579
--- /dev/null
+++ b/docs/CHANGELOG.md
@@ -0,0 +1,3 @@
+--8<-- "CHANGELOG.md"
+
+
diff --git a/docs/contributing.md b/docs/contributing.md
new file mode 100644
index 0000000..773a7e5
--- /dev/null
+++ b/docs/contributing.md
@@ -0,0 +1,3 @@
+--8<-- ".github/CONTRIBUTING.md"
+
+
diff --git a/docs/flowchart.md b/docs/flowchart.md
index ddac2e4..c5bde21 100644
--- a/docs/flowchart.md
+++ b/docs/flowchart.md
@@ -1,9 +1,5 @@
-# CHARLIE
-
-
-
-Flowchart
+# CHARLIE workflow

-> DISCLAIMER: This chart is for v0.8.x may be slightly outdated.
\ No newline at end of file
+> DISCLAIMER: This chart is for v0.8.x may be slightly outdated.
diff --git a/docs/index.md b/docs/index.md
index 409b166..0dc98bf 100644
--- a/docs/index.md
+++ b/docs/index.md
@@ -1,40 +1,3 @@
-## CHARLIE
-
-
-
-CHARLIE=**C**ircrnas in **H**ost **A**nd vi**R**uses ana**L**ysis p**I**p**E**line
-
-Things to know about CHARLIE:
-
-- Snakemake workflow to detect, annotate and quantify (DAQ) host and viral circular RNAs.
-- Primirarily developed to run on [BIOWULF](https://hpc.nih.gov/)
-- Reach out to [Vishal Koparde](mailto:vishal.koparde@nihgov) for questions/comments/requests.
-
-
-This circularRNA detection pipeline uses CIRCExplorer2, CIRI2 and many other tools in parallel to detect, quantify and annotate circRNAs. Here is a list of tools that can be run using CHARLIE:
-
-| circRNA Detection Tool | Aligner(s) | Run by default |
-| ---------------------- | ---------- | -------------- |
-| [CIRCExplorer2](https://github.com/YangLab/CIRCexplorer2) | STAR1 | Yes |
-| [CIRI2](https://sourceforge.net/projects/ciri/files/CIRI2/) | BWA1 | Yes |
-| [CIRCExplorer2](https://github.com/YangLab/CIRCexplorer2) | BWA1 | Yes |
-| [CLEAR](https://github.com/YangLab/CLEAR) | STAR1 | Yes |
-| [DCC](https://github.com/dieterich-lab/DCC) | STAR2 | Yes |
-| [circRNAFinder](https://github.com/bioxfu/circRNAFinder) | STAR3 | Yes |
-| [find_circ](https://github.com/marvin-jens/find_circ) | Bowtie2 | Yes |
-| [MapSplice](https://github.com/merckey/MapSplice2) | BWA2 | No |
-| [NCLScan](https://github.com/TreesLab/NCLscan) | NovoAlign | No |
-
-> Note: STAR1, STAR2, STAR3 denote 3 different sets of alignment parameters, etc.
-
-> Note: BWA1, BWA2 denote 2 different alignment parameters, etc.
-
-
-
+--8<-- "README.md"
+
diff --git a/docs/platforms.md b/docs/platforms.md
index 7633ed9..5ad72a9 100644
--- a/docs/platforms.md
+++ b/docs/platforms.md
@@ -1,19 +1,22 @@
-
CHARLIE was originally developed to run on biowulf, but it can run on other computing platforms too.
There are a few additional steps to configure CHARLIE.
+TODO
+
1. Clone CHARLIE.
- ```sh
- git clone https://github.com/CCBR/charlie
- ```
+
+ ```sh
+ git clone https://github.com/CCBR/charlie
+ ```
1. Initialize your project working directory.
- ```sh
- ```
+ ```sh
+
+ ```
1. Create a directory of reference files.
1. Edit your project's config file.
-1. If you are using a SLURM job scheduler, edit `cluster.json` and `submit_script.sbatch`.
\ No newline at end of file
+1. If you are using a SLURM job scheduler, edit `cluster.json` and `submit_script.sbatch`.
diff --git a/docs/references.md b/docs/references.md
index 4f169d4..f490a2c 100644
--- a/docs/references.md
+++ b/docs/references.md
@@ -1,12 +1,10 @@
-## CHARLIE
-
-
+## References
The reference sequences comprises of the host genome and the viral genomes.
### Fasta
-*hg38* and *mm39* genome builds are chosen to represent hosts. Ribosomal sequences (*45S, 5S*) are downloaded from NCBI. *hg38* and *mm39* were masked for rRNA sequence and *45S* and *5S* sequences from NCBI are appended as separate chromosomes. The following viral sequences were appended to the rRNA masked *hg38* reference:
+_hg38_ and _mm39_ genome builds are chosen to represent hosts. Ribosomal sequences (_45S, 5S_) are downloaded from NCBI. _hg38_ and _mm39_ were masked for rRNA sequence and _45S_ and _5S_ sequences from NCBI are appended as separate chromosomes. The following viral sequences were appended to the rRNA masked _hg38_ reference:
```bash
HOSTS:
@@ -31,6 +29,6 @@ VIRUSES:
* MH636806.1 [MHV68 (Murine herpesvirus 68 strain WUMS)]
```
-Location: The entire resource bundle is available at `/data/CCBR_Pipeliner/db/PipeDB/charlie/fastas_gtfs` on BIOWULF. This location also have additional bash scritpts required for aggregating annotations and building indices required by different aligners.
+Location: The entire resource bundle is available at `/data/CCBR_Pipeliner/db/PipeDB/charlie/fastas_gtfs` on BIOWULF. This location also have additional bash scripts required for aggregating annotations and building indices required by different aligners.
-When `-m=dryrun` is run for the first time after initialization (`-m=init`), the appropriate host+additives+viruses fasta and gtf files are created on the fly, which are then used to build aligner reference indexes automatically.
+When `-m=dryrun` is run for the first time after initialization (`-m=init`), the appropriate host+additives+viruses fasta and gtf files are created on the fly, which are then used to build aligner reference indexes automatically.
diff --git a/docs/requirements.txt b/docs/requirements.txt
new file mode 100644
index 0000000..083b20b
--- /dev/null
+++ b/docs/requirements.txt
@@ -0,0 +1,13 @@
+mkdocs
+#https://pypi.org/project/mkdocs-git-revision-date-localized-plugin/
+mkdocs-git-revision-date-localized-plugin
+#https://pypi.org/project/mkdocs-minify-plugin/
+mkdocs-minify-plugin
+#https://pypi.org/project/mkdocs-git-revision-date-plugin/
+mkdocs-git-revision-date-plugin
+#https://pypi.org/project/mkdocs-material/
+mkdocs-material
+#https://pypi.org/project/mkdocs-material-extensions/
+mkdocs-material-extensions
+#https://github.com/jimporter/mike
+mike
diff --git a/docs/tutorial.md b/docs/tutorial.md
index a2f3382..2b797c3 100644
--- a/docs/tutorial.md
+++ b/docs/tutorial.md
@@ -1,27 +1,22 @@
-# CHARLIE
-
-
-
-### Tutorial
+# Tutorial
#### Prerequisites
-* [Biowulf](https://hpc.nih.gov/) account: Biowulf account can be requested [here](https://hpc.nih.gov/docs/accounts.html).
+- [Biowulf](https://hpc.nih.gov/) account: Biowulf account can be requested [here](https://hpc.nih.gov/docs/accounts.html).
-* Membership to Ziegelbauer user group on Biowulf. You can check this by typing the following command:
+- Membership to Ziegelbauer user group on Biowulf. You can check this by typing the following command:
```bash
% groups
```
-
output:
- ```bash
- CCBR kopardevn Ziegelbauer_lab
- ```
+```bash
+CCBR kopardevn Ziegelbauer_lab
+```
- If `Ziegelbauer_lab` is not listed then you can email a request to be added to the groups [here](mailto:staff@hpc.nih.gov)
+If `Ziegelbauer_lab` is not listed then you can email a request to be added to the groups [here](mailto:staff@hpc.nih.gov)
#### Location
@@ -138,9 +133,9 @@ Optional Arguments:
Example commands:
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/ouput/folder -m=init
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/ouput/folder -m=dryrun
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/ouput/folder -m=run
+ bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=init
+ bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=dryrun
+ bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=run
##########################################################################################
@@ -153,8 +148,8 @@ VersionInfo:
##########################################################################################
```
->NOTE:
->You can replace `v0.10.0` in the above command with the latest version to use a newer version. `run_circrna_daq.sh` was called `test.sh` in versions older than `v0.4.0`.
+> NOTE:
+> You can replace `v0.10.0` in the above command with the latest version to use a newer version. `run_circrna_daq.sh` was called `test.sh` in versions older than `v0.4.0`.
To initial the working directory run:
@@ -174,16 +169,16 @@ The above command creates `` folder and creates 2 subfolders
This file is used to fine tune the execution of the pipeline by setting:
-* sample sheet location ... aka `samples.tsv`
-* the temporary directory -- make sure this is correct for your computing environment.
-* which circRNA finding tools to use by editing these:
- * run_clear: True
- * run_dcc: True
- * run_mapsplice: False
- * run_circRNAFinder: True
- * run_nclscan: False
- * run_findcirc: False
-* describes the location of other resources/indexes/tools etc. Generally, these do NOT need to be changed.
+- sample sheet location ... aka `samples.tsv`
+- the temporary directory -- make sure this is correct for your computing environment.
+- which circRNA finding tools to use by editing these:
+ - run_clear: True
+ - run_dcc: True
+ - run_mapsplice: False
+ - run_circRNAFinder: True
+ - run_nclscan: False
+ - run_findcirc: False
+- describes the location of other resources/indexes/tools etc. Generally, these do NOT need to be changed.
##### samples.tsv
@@ -312,7 +307,7 @@ output:
├── logs/snakemake.log.jobby.short
├── logs/snakemake.log.jobby.txt
├── logs
-│ ... log files ...
+│ ... log files ...
│ ... skipping ...
├── nclscan.config
├── qc
@@ -1005,47 +1000,46 @@ grep FAIL logs/snakemake.log.jobby.short
The above command also gives `.err` and `.out` log files which can give further insights on reasons for failure and changes required to be made for a successful run.
-
#### Expected output:
The main output file is `results/circRNA_master_counts.tsv.gz`. Here are the top 3 tiles from an example output:
-| Column_number | Column_title | Example_1 | Example_2 | Example_3 |
-| ------------- | ---------------------------------------------- | ---------- | ---------- | ---------- |
-| 1 | chrom | GL000220.1 | GL000220.1 | GL000220.1 |
-| 2 | start | 107635 | 112482 | 118578 |
-| 3 | end | 151634 | 156427 | 118759 |
-| 4 | circExplorer_strand | \-1 | \-1 | \-1 |
-| 5 | circExplorer_bwa_strand | . | . | . |
-| 6 | ciri_strand | \-1 | \-1 | \-1 |
-| 7 | dcc_strand | \-1 | \-1 | \-1 |
-| 8 | circrnafinder_strand | \-1 | \-1 | \-1 |
-| 9 | flanking_sites_+ | CC##GC | GC##CC | CC##GC |
-| 10 | flanking_sites_- | GG##GC | GC##GG | GG##GC |
-| 11 | sample_name | GI1_N | GI1_N | GI1_N |
-| 12 | ntools | 1 | 1 | 1 |
-| 13 | HQ | N | N | N |
-| 14 | circExplorer_read_count | \-1 | \-1 | \-1 |
-| 15 | circExplorer_found_BSJcounts | \-1 | \-1 | \-1 |
-| 16 | circExplorer_found_linear_BSJ_+_counts | \-1 | \-1 | \-1 |
-| 17 | circExplorer_found_linear_spliced_BSJ_+_counts | \-1 | \-1 | \-1 |
-| 18 | circExplorer_found_linear_BSJ_-_counts | \-1 | \-1 | \-1 |
-| 19 | circExplorer_found_linear_spliced_BSJ_-_counts | \-1 | \-1 | \-1 |
-| 20 | circExplorer_found_linear_BSJ_._counts | \-1 | \-1 | \-1 |
-| 21 | circExplorer_found_linear_spliced_BSJ_._counts | \-1 | \-1 | \-1 |
-| 22 | ciri_read_count | \-1 | \-1 | \-1 |
-| 23 | ciri_linear_read_count | \-1 | \-1 | \-1 |
-| 24 | circExplorer_bwa_read_count | 3 | 7 | 3 |
-| 25 | dcc_read_count | \-1 | \-1 | \-1 |
-| 26 | dcc_linear_read_count | \-1 | \-1 | \-1 |
-| 27 | circrnafinder_read_count | \-1 | \-1 | \-1 |
-| 28 | hqcounts | 1 | 1 | 1 |
-| 29 | nonhqcounts | 0 | 0 | 0 |
-| 30 | circExplorer_annotation | Unknown | Unknown | Unknown |
-| 31 | ciri_annotation | Unknown | Unknown | Unknown |
-| 32 | circExplorer_bwa_annotation | novel | novel | novel |
-| 33 | dcc_gene | Unknown | Unknown | Unknown |
-| 34 | dcc_junction_type | Unknown | Unknown | Unknown |
-| 35 | dcc_annotation | Unknown | Unknown | Unknown |
-
-Expected output from the sample data is stored under `.tests/expected_output`.
\ No newline at end of file
+| Column_number | Column_title | Example_1 | Example_2 | Example_3 |
+| ------------- | ----------------------------------------------- | ---------- | ---------- | ---------- |
+| 1 | chrom | GL000220.1 | GL000220.1 | GL000220.1 |
+| 2 | start | 107635 | 112482 | 118578 |
+| 3 | end | 151634 | 156427 | 118759 |
+| 4 | circExplorer_strand | \-1 | \-1 | \-1 |
+| 5 | circExplorer_bwa_strand | . | . | . |
+| 6 | ciri_strand | \-1 | \-1 | \-1 |
+| 7 | dcc_strand | \-1 | \-1 | \-1 |
+| 8 | circrnafinder_strand | \-1 | \-1 | \-1 |
+| 9 | flanking*sites*+ | CC##GC | GC##CC | CC##GC |
+| 10 | flanking*sites*- | GG##GC | GC##GG | GG##GC |
+| 11 | sample_name | GI1_N | GI1_N | GI1_N |
+| 12 | ntools | 1 | 1 | 1 |
+| 13 | HQ | N | N | N |
+| 14 | circExplorer_read_count | \-1 | \-1 | \-1 |
+| 15 | circExplorer_found_BSJcounts | \-1 | \-1 | \-1 |
+| 16 | circExplorer*found_linear_BSJ*+\_counts | \-1 | \-1 | \-1 |
+| 17 | circExplorer*found_linear_spliced_BSJ*+\_counts | \-1 | \-1 | \-1 |
+| 18 | circExplorer*found_linear_BSJ*-\_counts | \-1 | \-1 | \-1 |
+| 19 | circExplorer*found_linear_spliced_BSJ*-\_counts | \-1 | \-1 | \-1 |
+| 20 | circExplorer*found_linear_BSJ*.\_counts | \-1 | \-1 | \-1 |
+| 21 | circExplorer*found_linear_spliced_BSJ*.\_counts | \-1 | \-1 | \-1 |
+| 22 | ciri_read_count | \-1 | \-1 | \-1 |
+| 23 | ciri_linear_read_count | \-1 | \-1 | \-1 |
+| 24 | circExplorer_bwa_read_count | 3 | 7 | 3 |
+| 25 | dcc_read_count | \-1 | \-1 | \-1 |
+| 26 | dcc_linear_read_count | \-1 | \-1 | \-1 |
+| 27 | circrnafinder_read_count | \-1 | \-1 | \-1 |
+| 28 | hqcounts | 1 | 1 | 1 |
+| 29 | nonhqcounts | 0 | 0 | 0 |
+| 30 | circExplorer_annotation | Unknown | Unknown | Unknown |
+| 31 | ciri_annotation | Unknown | Unknown | Unknown |
+| 32 | circExplorer_bwa_annotation | novel | novel | novel |
+| 33 | dcc_gene | Unknown | Unknown | Unknown |
+| 34 | dcc_junction_type | Unknown | Unknown | Unknown |
+| 35 | dcc_annotation | Unknown | Unknown | Unknown |
+
+Expected output from the sample data is stored under `.tests/expected_output`.
diff --git a/docs/versions.md b/docs/versions.md
deleted file mode 100644
index e5e1919..0000000
--- a/docs/versions.md
+++ /dev/null
@@ -1,89 +0,0 @@
-# CHARLIE
-
-
-
-### Version/Release highlights
-
-#### v0.1.0
-
-* base version
-* PE only support
-
-#### v0.2.x
-
-* SE support added .. PE/SE samples handled concurrently
-* `envmodules` used in Snakemake in place of `module load` statements
-
-#### v0.3.x
-
-* Lookup table for hg38 to hg19 circRNA annotations is updated... this eliminate one-to-many hits from the previous version
-* BSJs extracted as different bam file.
-* flowchart added
-* adding slurmjobid to log/err file names
-* v0.3.1 has significant (>10X) performance improvements at BSJ bam creation
-* v0.3.3 splits BSJ bams into human and viral bams, and also converts them to bigwigs
-* v0.3.4 adds hg38_rRNA_masked_plus_rRNA_plus_viruses_plus_ERCC reference (source:Sarah)
-
-#### v0.4.x
-
-* [CLEAR](https://github.com/YangLab/CLEAR) added.
-* wrapper script (`run_circrna_daq.sh`) added for local and cluster execution.
-
-* "spliced reads only" bam created and split by regions
-
-#### v0.5.x
-
-* `run_clear` is now set to True (as default)
-* `circ_quant` replaces `clear_quant` in the CLEAR rule. In order words, we are reusing the STAR alignment file and the circExplorer2 output file for running CLEAR. No need to run HISAT2 and TopHat (fusion-search with Bowtie1). This is much quicker.
-* Using picard to estimate duplicates using[ *MarkDuplicates*](https://gatk.broadinstitute.org/hc/en-us/articles/360037052812-MarkDuplicates-Picard-)
-* Generating a per-run [multiqc](https://multiqc.info/) HTML report
-* Using [*eulerr*](https://www.rdocumentation.org/packages/eulerr/versions/6.1.0) R package to generate CIRI-CircExplorer circRNA Venn diagrams and include them in the mulitqc report
-* Gather per job cluster metadata like queue time, run time, job state etc. Stats are compiled in **HPC_summary** file
-* CLEAR pipeline *quant.txt* file is annotated for known circRNAs
-* `WORKDIR` can now be a relative path
-* bam2bw conversion fix for BSJ and spliced_reads. [Issue](https://github.com/kopardev/circRNA/issues/17) closed!
-
-
-#### v0.6.x
-
-* cutadapt_min_length to cutadapt rule... setting it to 15 in config.
-* customBSJs recalled from STAR alignments
- * only for PE
- * removes erroneously called CircExplorer BSJs
-* create sense and anti-sense BSJ BAMs and BW for each reference (host+viruses)
-* find reads which contribute to CIRI BSJs but not on the STAR list of BSJ reads, see if they contribute to novel (not called by STAR) BSJs and append novel BSJs to customBSJ list
-* create linear reads BAM file
-* create linear reads BigWigs for each region in the .regions file.
-* Optimized pysam scripts
-* fixed premature completion of singularity rules
-
-#### v0.7.x
-
-* 5 circRNA callers
-* all-sample counts matrix with annotations
-
-#### v0.9.x
-
-* updates to wrapper script, many new arguments/options added
-* new per-sample counts table format
-* new all-sample master counts matrix with min-nreads filtering and ntools column to show number of tools supporting the circRNA call
-* new version of Snakemake
-* cluster_status script added for forced completion of pipeline upon TIMEOUTs
-updated flowchart from lucid charts
-* added circRNAfinder, find_circ, circExplorer2_bwa and other tools
-* optimized execution and resource requirements
-* updated viral annotations (Thanks Sara!)
-* new method to extract linear counts, create linear BAMs using circExplorer2 outputs
-* new job reporting using jobby and its derivatives
-separated creation of BWA and BOWTIE2 index from creation of STAR index to speed things up
-* parallelized find_circ
-better cleanup (eg. deleting _STARgenome folders, etc.) for much smaller digital footprint
-* multitude of comments throughout the snakefiles including listing of output file column descriptions
-* preliminary GH actions added
-
-#### v0.10.x
-
-* strand are reported together, strand from all callers are reported,
-* both + and - flanking sites are reported,
-* rev-comp function updated,
-* updated versions of tools to match available tools on BIOWULF.
\ No newline at end of file
diff --git a/mkdocs.yml b/mkdocs.yml
index a405628..dcd9d9f 100644
--- a/mkdocs.yml
+++ b/mkdocs.yml
@@ -4,18 +4,42 @@ site_author: Vishal Koparde, Ph.D.
# Repository
repo_name: CCBR/CHARLIE
repo_url: https://github.com/CCBR/CHARLIE
+edit_uri: https://github.com/CCBR/CHARLIE/edit/main
# Copyright
-copyright: Copyright © 2023 CCBR
+copyright: Copyright © 2023 CCR Collaborative Bioinformatics Resource
# Configuration
theme:
name: material
+ features:
+ - navigation.tabs
+ - navigation.top
+ - navigation.indexes
+ - toc.integrate
palette:
- scheme: default
+ - scheme: default
+ primary: indigo
+ accent: indigo
+ toggle:
+ icon: material/toggle-switch-off-outline
+ name: Switch to dark mode
+ - scheme: slate
+ primary: red
+ accent: red
+ toggle:
+ icon: material/toggle-switch
+ name: Switch to light mode
plugins:
- search
+ - git-revision-date
+ - minify:
+ minify_html: true
+ - mike:
+ alias_type: symlink
+ canonical_version: latest
+ version_selector: true
# Customization
extra:
@@ -26,11 +50,48 @@ extra:
link: https://github.com/CCBR
- icon: fontawesome/brands/docker
link: https://hub.docker.com/orgs/nciccbr/repositories
+ version:
+ provider: mike
+
+markdown_extensions:
+ - markdown.extensions.admonition
+ - markdown.extensions.attr_list
+ - markdown.extensions.md_in_html
+ - markdown.extensions.def_list
+ - markdown.extensions.footnotes
+ - markdown.extensions.meta
+ - markdown.extensions.toc:
+ permalink: true
+ - pymdownx.arithmatex:
+ generic: true
+ - pymdownx.caret
+ - pymdownx.critic
+ - pymdownx.details
+ - pymdownx.emoji:
+ emoji_index: !!python/name:materialx.emoji.twemoji
+ emoji_generator: !!python/name:materialx.emoji.to_svg
+ - pymdownx.highlight
+ - pymdownx.inlinehilite
+ - pymdownx.keys
+ - pymdownx.magiclink:
+ repo_url_shorthand: true
+ user: squidfunk
+ repo: mkdocs-material
+ - pymdownx.mark
+ - pymdownx.smartsymbols
+ - pymdownx.snippets:
+ check_paths: true
+ - pymdownx.superfences
+ - pymdownx.tabbed
+ - pymdownx.tasklist:
+ custom_checkbox: true
+ - pymdownx.tilde
# Page Tree
nav:
- Home: index.md
- - Flowchart: flowchart.md
+ - Workflow: flowchart.md
- Tutorial: tutorial.md
- References: references.md
- - Versions: versions.md
+ - Changelog: CHANGELOG.md
+ - How to contribute: contributing.md