The selection of human leukocyte antigen (HLA) epitopes is critical in development of vaccine and immunotherapy strategies. Recent strides in liquid chromatography and mass spectrometry have expedited the in-depth characterization of the HLA-presented ligandome. Concurrently with these technological advancements, the development of efficient methods for deciphering immunopeptidomics data and robust (neo)antigen presentation predictors is urgently needed with vast potential. Here, we developed the ImmuneApp, which facilitates prediction of antigen presentation, scoring for neoepitope immunogenicity, and immunopeptidomics analysis, with enhanced precision. ImmuneApp harnesses an interpretable, attention-based hybrid deep learning framework for predicting HLA-I epitopes trained on 349,650 ligands, enabling the extraction of informative embeddings and identification of critical residues involved in mediating pHLA binding specificity. Evaluation conducted on independent mono-allelic dataset demonstrated that ImmuneApp significantly outperforms existing methods for antigen presentation prediction. Additionally, we present a more accurate model-based deconvolution approach and conduct a systematic analysis of 216 publicly available multi-allelic immunopeptidomics samples, resulting in the deconvolution of 835,551 ligands restricted to over 100 distinct HLA-I alleles. Our investigation highlights the effectiveness of a composite model, denoted as ImmuneApp-MA, which integrates both mono- and multi-allelic data modalities to enhance predictive performance. Leveraging ImmuneApp-MA as a pre-trained model for deep transfer learning on a curated immunogenicity dataset, we introduce ImmuneApp-Neo, a novel immunogenicity predictor that outperforms existing state-of-the-art methods in prioritizing immunogenic neoepitopes, yielding a notable 2.1-fold improvement in positive predictive value (PPV). We further demonstrate the utility of ImmuneApp across diverse disease-related immunopeptidomics datasets sourced from tumor tissues and cancer biopsies, highlighting its efficacy in various tasks including quality control, binding annotations, HLA assignment, motif discovery and elucidation, and antigen presentation prediction on a sample-specific manner.
Download ImmuneApp by
git clone https://github.com/bsml320/ImmuneApp
Installation has been tested in Linux server, CentOS Linux release 7.8.2003 (Core), with Python 3.7. Since the package is written in python 3x, python3x with the pip tool must be installed. ImmuneApp uses the following dependencies: numpy, scipy, pandas, h5py, keras version=2.3.1, tensorflow=1.15, seaborn, logomaker, and shutil, pathlib. We highly recommend that users leave a message under the ImmuneApp issue interface (https://github.com/bsml320/ImmuneApp/issues) when encountering any installation and running problems. We will deal with it in time. You can install these packages by the following commands:
conda create -n ImmuneApp python=3.7
conda activate ImmuneApp
pip install numpy==1.20.0
pip install pandas==1.3.3
pip install scipy==1.7.1
pip install keras==2.3.1
pip install tensorflow==1.15
pip install seaborn==0.11.2
pip install logomaker==0.8
pip install pathlib
pip install protobuf==3.20
pip install h5py==2.10.0
Note: please unzip the "gibbscluster-2.0f.Linux.tar.gz", and make sure the user has the read and execute permission on the gibbscluster program.
cd ImmuneApp/
tar -zxvf gibbscluster-2.0f.Linux.tar.gz
Once the file is unzipped, the user gets the gibbscluster-2.0 software. Modify line 14 of the “gibbscluster” file in the gibbscluster-2.0 directory to the full path gibbscluster-2.0 software - Set FULL PATH to the GibbsCluster 2.0 directory (mandatory).
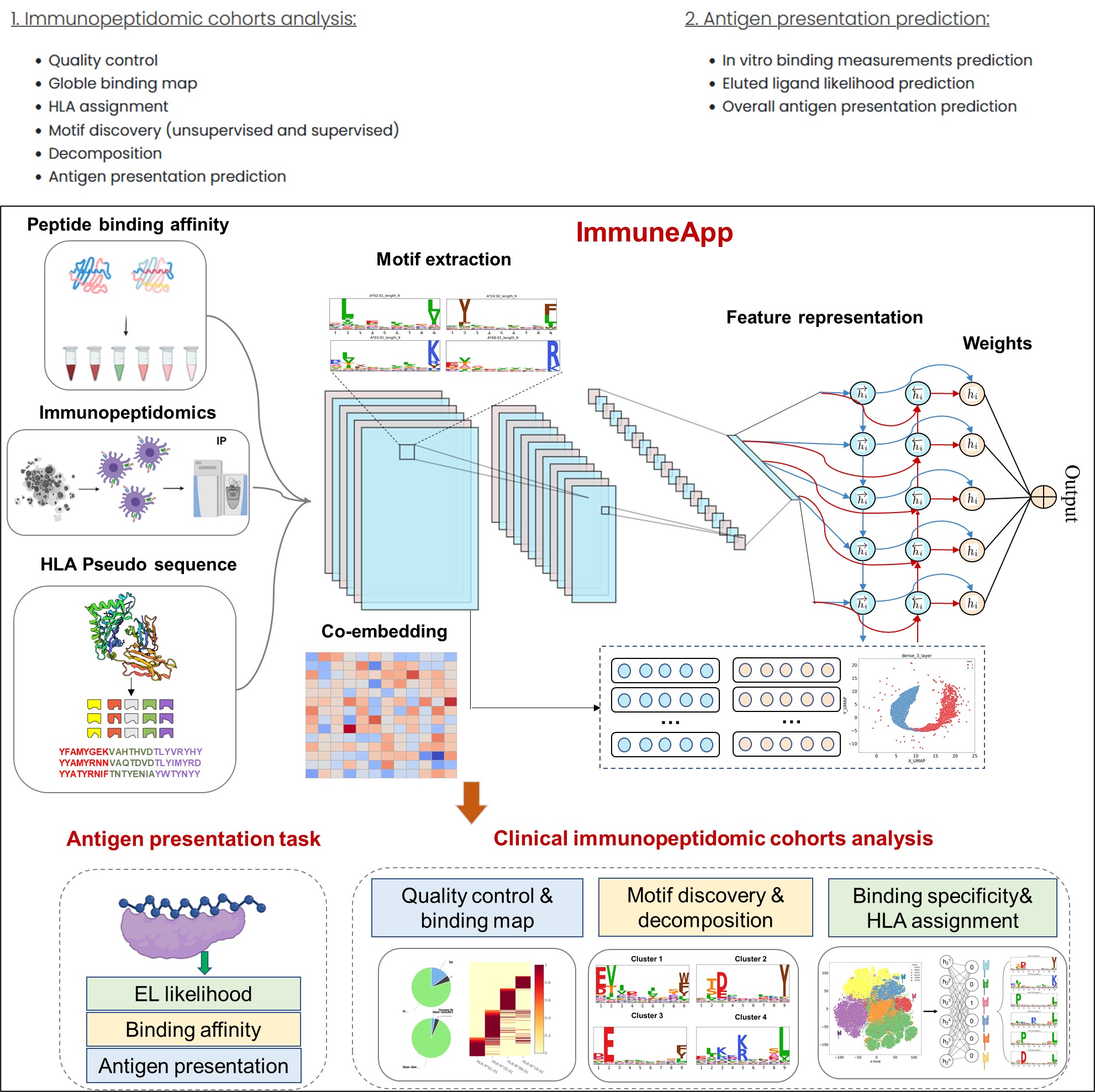
ImmuneApp provides three services: prediction of antigen presentation, scoring for neoepitope immunogenicity, and immunopeptidomics analysis, with enhanced precision.
1. For antigen presentation prediction, this module accept two different types of input; FASTA and Peptide. In addition, candidate HLA molecules should be specified in the predictions. For FASTA input, the peptide length(s) should be specified.
For peptides input, please uses:
cd ImmuneApp/
python ImmuneApp_presentation_prediction.py -f 'testdata/test_peplist.txt' -a 'HLA-A*01:01' 'HLA-A*02:01' 'HLA-A*03:01' 'HLA-B*07:02' -b -o 'results'
For FASTA input, please uses:
python ImmuneApp_presentation_prediction.py -fa 'testdata/test.fasta' -a 'HLA-A*01:01' 'HLA-A*02:01' 'HLA-A*03:01' 'HLA-B*07:02' -b -o 'results'
2. For immunopeptidome analysis, this module accept immunopeptidomic samples as input, together with HLA molecule(s) by HLA tying tool.
For single sample, please uses:
python ImmuneApp_immunopeptidomics_analysis.py -f testdata/Melanoma_tissue_sample_of_patient_5.txt -a HLA-A*01:01,HLA-A*25:01,HLA-B*08:01,HLA-B*18:01 -o results
For multiple samples, separate the different sample names or HLA alleles with spaces, uses:
python ImmuneApp_immunopeptidomics_analysis.py -f testdata/Melanoma_tissue_sample_of_patient_5.txt testdata/Melanoma_tissue_sample_of_patient_8.txt -a HLA-A*01:01,HLA-A*25:01,HLA-B*08:01,HLA-B*18:01 HLA-A*01:01,HLA-A*03:01,HLA-B*07:02,HLA-B*08:01,HLA-C*07:02,HLA-C*07:01 -o results
3. For scoring for neoepitope immunogenicity, this module accept peptides as input, together with HLA molecule(s).
For peptides input, please uses:
python ImmuneApp_immunogenicity_prediction.py -f testdata/test_immunogenicity.txt -a 'HLA-A*01:01' 'HLA-A*02:01' 'HLA-A*03:01' 'HLA-B*07:02' -o results
For details of other parameters, run:
python ImmuneApp_immunopeptidomics_analysis.py --help
python ImmuneApp_presentation_prediction.py --help
python ImmuneApp_immunogenicity_prediction.py --help
Researchers can run ImmuneApp online at https://bioinfo.uth.edu/iapp/. For commercial usage inquiries, please contact the authors.
ImmuneApp implements four main modules: “Discovery”, “Analysis”, “Results” and “Controller”. In the backend, three well-trained deep learning models (ImmuneApp_BA, ImmuneApp_EL and ImmuneApp_AP) are used for the predictions of binding affinities, ligand probabilities, and overall antigen presentation as well as immunopeptidomic cohorts analysis, respectively. The “Controller” module checks the input data format, sends the data from frontend interfaces to the backend, creates the results using models, and then provides the results on the “Results” page. We implemented both pages in a responsive manner by using the HTML5, CSS, Bootstrap3, and JavaScript. Additionally, the "Controller" is called through Ajax technology to submit jobs, retrieve data, and show results. There is no limit to the number of tasks submitted by each user. ImmuneApp can automatically handle the jobs in a queue, which allows up to 5 jobs to execute concurrently.
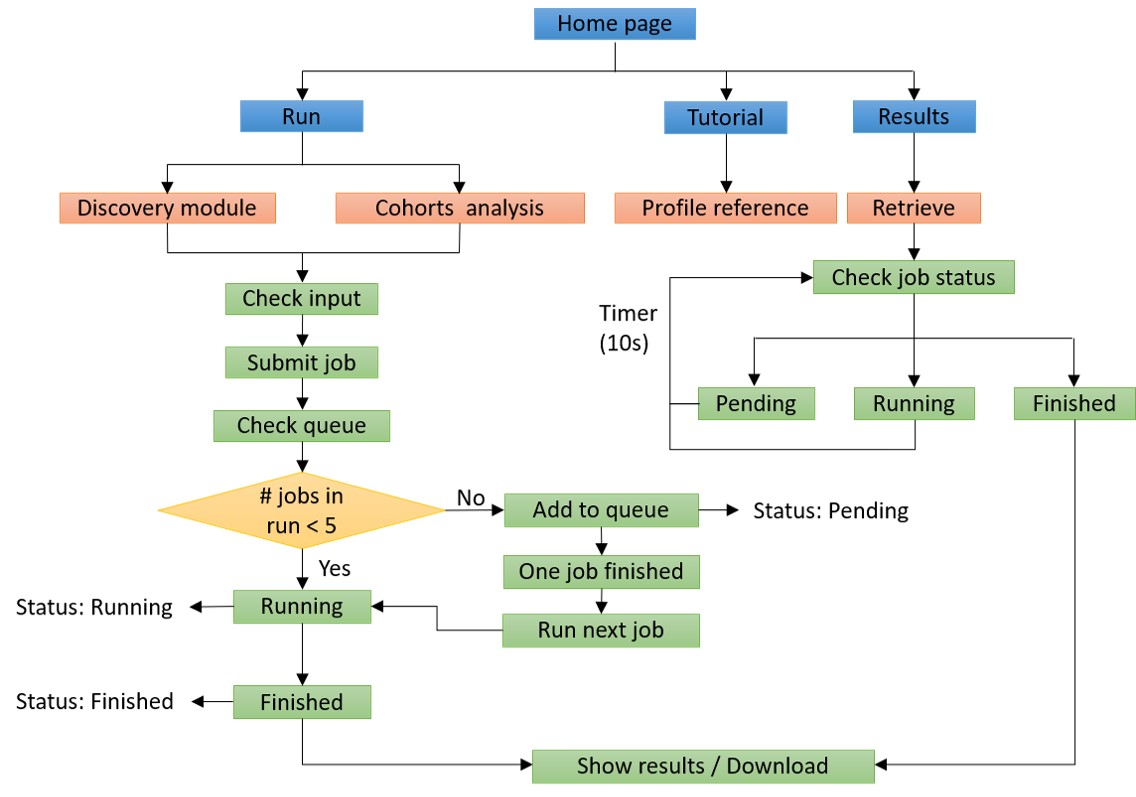
The “Discovery” module accepts two input types: “FASTA” and “Peptide”. Users can directly copy the input data to an online submission text box. Moreover, MHC molecules and the peptide length (only FASTA input) need to be specified for running prediction. The “Analysis” module accepts clinical immunopeptidomic samples as input, together with MHC molecules. The input sample(s) can be directly copied to an online submission text box or uploaded from the users local disk. Sample identity should be specified. This module provides intuitive report for personalized analysis, statistical reports, and visualization of results for clinical immunopeptidomic cohorts.
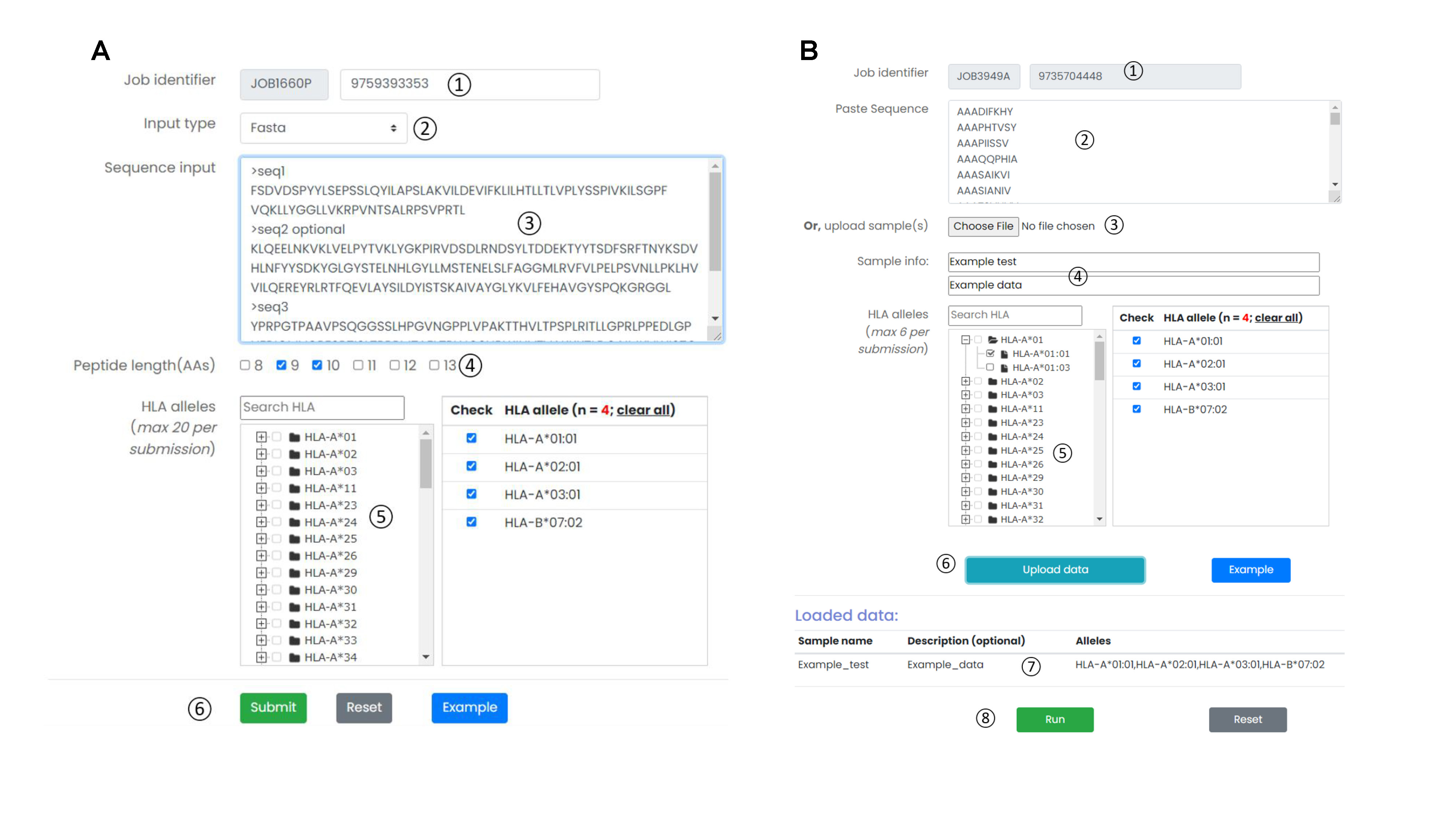
- Job identifier: Job identifier can be generated automatically or customized by the submitter. It is confidential to other users and can be used for job status monitoring and result retrieval.(See Results page).It is required.
- Input type: Provides two input formats, including the classic protein FASTA format and direct input of multiple peptides.
- Input textarea: The user can directly copy the protein sequence or peptide data in the input box.
- Peptide length(AAs): When the input method is Fasta format. The user needs to select one or more peptide lengths so that the server can construct a library of candidate antigen peptides.
- HLA alleles: The ImmuneApp 1.0 server predicts peptides binding to more than 10,000 human MHC molecule. We constructed a classification tree of HLA. Users can quickly retrieve and submit candidate HLA alleles through the search box and tree map. Each submitted task is allowed to select up to 20 HLA alleles.
- Operation buttons: Submit, reset the submission form, or access the example dataset.
- Job identifier: Job identifier can be generated automatically or customized by the submitter. It is confidential to other users and can be used for job status monitoring and result retrieval.(See Results page).It is required.
- Input textarea: The user can directly copy immunopeptidomic cohorts sample data in the input box.
- Upload sample(s): The user can also upload immunopeptidomic cohorts sample to the server.
- Sample info: The user needs to provide identification information for each sample.
- HLA alleles: The ImmuneApp 1.0 server predicts peptides binding to more than 10,000 human MHC molecule. We constructed a classification tree of HLA. Users can quickly retrieve and submit candidate HLA alleles through the search box and tree map. Each submitted task is allowed to select up to 6 HLA alleles.
- Operation buttons: Upload immunopeptidomic cohorts sample to the server by this button
- loaded data: A list of immunopeptidomic cohorts uploaded by users for analysis.
- Operation buttons: Submit, reset the submission form, or access the example dataset.
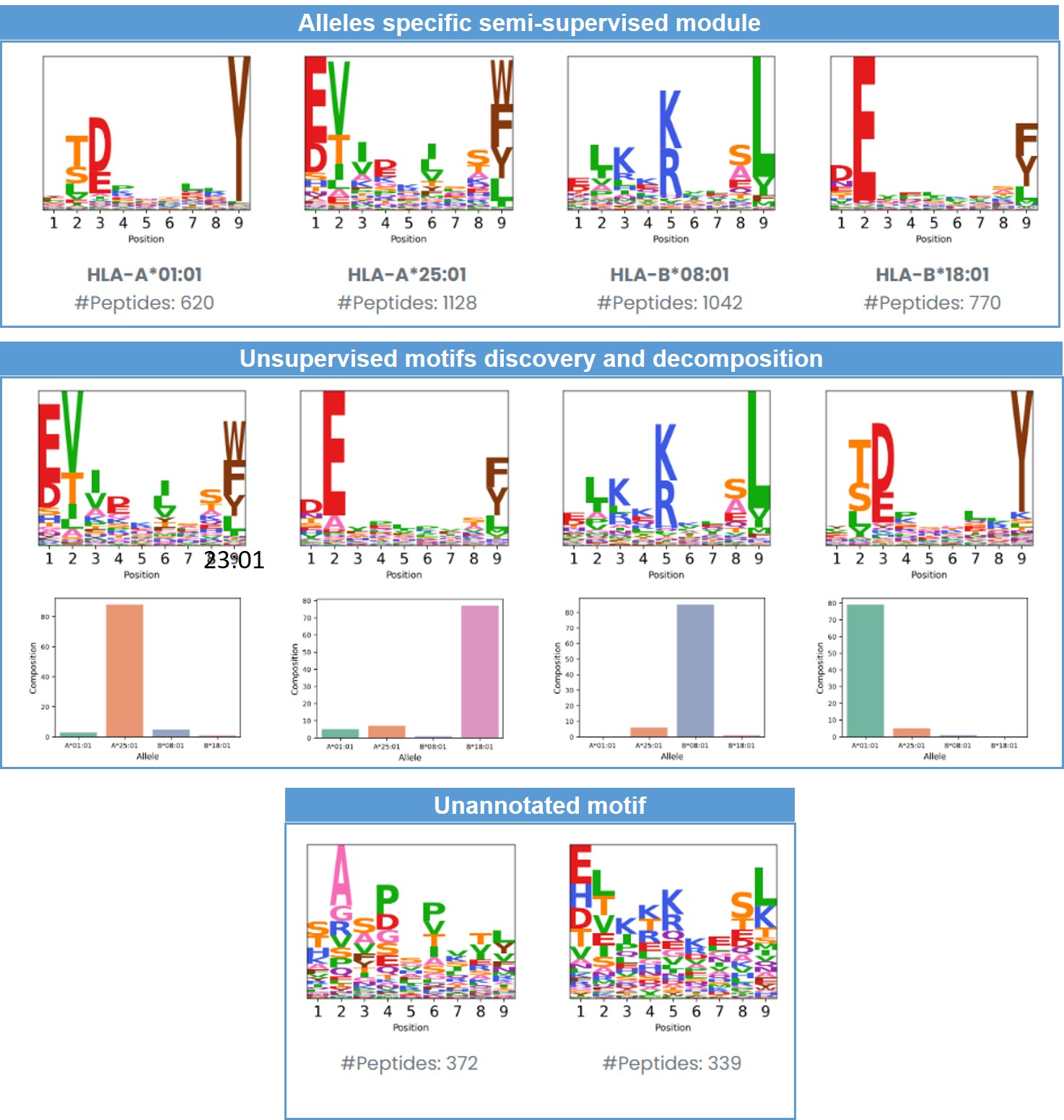
- Analysis, statistics, and visualization for melanoma-associated samples using ImmuneApp.

- Motif discovery and decomposition for melanoma-associated samples using ImmuneApp: