FUSION Benchmarking
library(dplyr)
library(ggplot2)
library(tidyr)
library(stringr)Attaching package: ‘dplyr’
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
These benchmarking data come from the following paper: https://pubmed.ncbi.nlm.nih.gov/31639029/ and resource: https://data.broadinstitute.org/Trinity/STAR_FUSION_PAPER/SupplementaryData/sim_reads/.
Sim1 and Sim5 were excluded, as Squid did not sufficiently process these samples. This is due to a bug within the Squid algorithm, and is likely due to the simulated nature of the data.
Fusion calls with 2 or more callers supporting the call were used in the comparison below.
fusion_truth_set <- read.delim('/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/FUSION/sim_101.truth_set.dat', sep="|", header=F)
colnames(fusion_truth_set) <- c('sampleID', 'fusion')
fusion_truth_set <- fusion_truth_set %>% dplyr::filter(sampleID %in% c('sim2_reads', 'sim3_reads', 'sim4_reads'))# Give the input file name to the function.
sim2 <- read.delim("/projects/compsci/omics_share/meta/benchmarking/FUSION_human_nxf_bench/sim2_reads/fusion-report/sim2_reads_fusion_list_filtered.tsv", sep = '\t', header = F, col.names = 'fusion')
sim2$sampleID <- 'sim2_reads'
sim3 <- read.delim("/projects/compsci/omics_share/meta/benchmarking/FUSION_human_nxf_bench/sim3_reads/fusion-report/sim3_reads_fusion_list_filtered.tsv", sep = '\t', header = F, col.names = 'fusion')
sim3$sampleID <- 'sim3_reads'
sim4 <- read.delim("/projects/compsci/omics_share/meta/benchmarking/FUSION_human_nxf_bench/sim4_reads/fusion-report/sim4_reads_fusion_list_filtered.tsv", sep = '\t', header = F, col.names = 'fusion')
sim4$sampleID <- 'sim4_reads'joined_truth_test_sim2 <- dplyr::full_join((fusion_truth_set %>% dplyr::filter(sampleID == 'sim2_reads')), sim2, by = 'fusion')
joined_truth_test_sim3 <- dplyr::full_join((fusion_truth_set %>% dplyr::filter(sampleID == 'sim3_reads')), sim3, by = 'fusion')
joined_truth_test_sim4 <- dplyr::full_join((fusion_truth_set %>% dplyr::filter(sampleID == 'sim4_reads')), sim4, by = 'fusion')all_joined_test_sim <- do.call("rbind", list(joined_truth_test_sim2, joined_truth_test_sim3, joined_truth_test_sim4))venn_input <- all_joined_test_sim %>% mutate(Truth_Set = ifelse(is.na(sampleID.x), 'FALSE', 'TRUE'),
Test_Set = ifelse(is.na(sampleID.y), 'FALSE', 'TRUE')) %>%
dplyr::select(Truth_Set, Test_Set)
venn_input$Truth_Set <- as.logical(venn_input$Truth_Set)
venn_input$Test_Set <- as.logical(venn_input$Test_Set)
library("ggvenn")
ggvenn(data = venn_input) Loading required package: grid
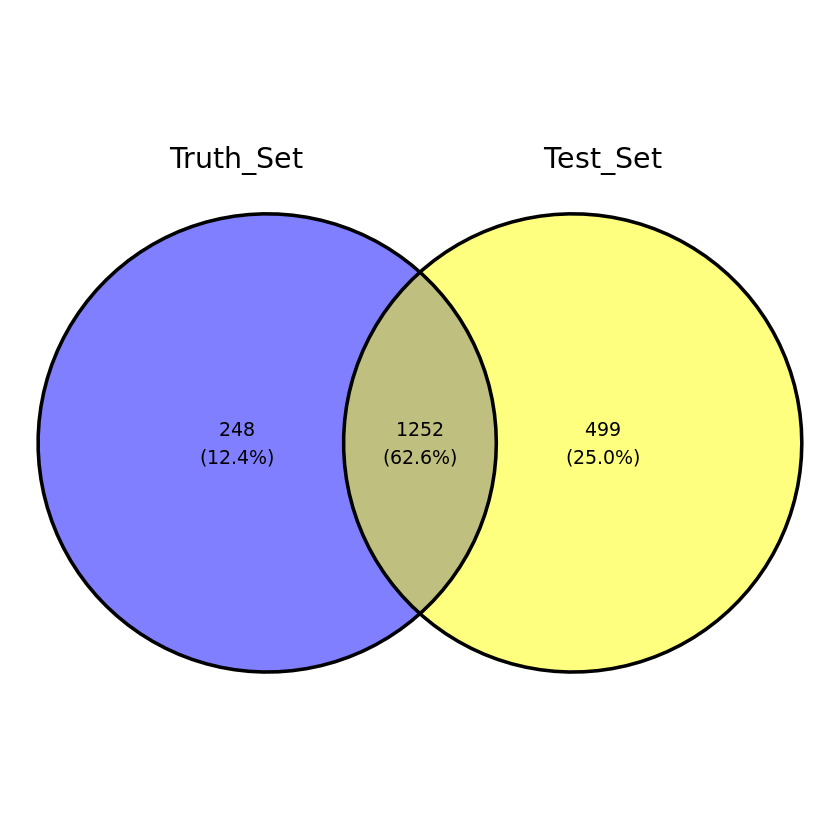
The test data were generated from a GTF source that included lincRNA and other transcript sources that were filtered by CTAT in their reference build. A 1:1 overlap should not be expected