RNA Benchmarking
What follows is text from an R based Jupyter notebook that generate simulated RNA data. The output from the pipeline is then compared against truth.
The Jupyter notebook is found here: /projects/omics_share/meta/scripts/benchmarking
#BiocManager::install("Rsubread")
# https://rdrr.io/bioc/Rsubread/man/simReads.htmllibrary(Rsubread)
library(dplyr)
library(ggplot2)
library(tidyr)
library(stringr)Attaching package: ‘dplyr’
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
Files used:
wget http://ftp.ensembl.org/pub/release-102/fasta/mus_musculus/cds/Mus_musculus.GRCm38.cds.all.fa.gz
gunzip Mus_musculus.GRCm38.cds.all.fa.gz
reformat.sh in=Mus_musculus.GRCm38.cds.all.fa out=Mus_musculus.GRCm38.cds.all.LINEWRAP.fa fastawrap=80
BBMap is the source of reformat.sh. This script takes a single line fasta and turns it to 80 character line wrapped fasta.
simReads can't use single line FASTA. An error is thrown.
# Scan through the fasta file to get transcript names and lengths
transcripts <- scanFasta('/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus.GRCm38.cds.all.LINEWRAP.fa')
nsequences <- nrow(transcripts) - sum(transcripts$Duplicate)
Warning: 17539 duplicate sequences were found in the input. Please check the summary table.
head(transcripts)
#transcripts %>% filter(Duplicated == TRUE)
| TranscriptID | Length | MD5 | Unique | Occurrence | Duplicated | |
|---|---|---|---|---|---|---|
| <chr> | <int> | <chr> | <lgl> | <int> | <lgl> | |
| 1 | ENSMUST00000177564.1 cds chromosome:GRCm38:14:54122226:54122241:1 gene:ENSMUSG00000096176.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:Trdd2 description:T cell receptor delta diversity 2 [Source:MGI Symbol;Acc:MGI:4439546] | 16 | 587E802B733230F3DA2F88F6EB24F77E | TRUE | 1 | FALSE |
| 2 | ENSMUST00000196221.1 cds chromosome:GRCm38:14:54113468:54113476:1 gene:ENSMUSG00000096749.2 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:Trdd1 description:T cell receptor delta diversity 1 [Source:MGI Symbol;Acc:MGI:4439547] | 9 | E694792CBB1DF84D2D6CCBE57D8F3C66 | TRUE | 1 | FALSE |
| 3 | ENSMUST00000178537.1 cds chromosome:GRCm38:6:41533201:41533212:1 gene:ENSMUSG00000095668.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:Trbd1 description:T cell receptor beta, D region 1 [Source:MGI Symbol;Acc:MGI:4439571] | 12 | 1F47B55923E2D23090F894C439974B55 | TRUE | 1 | FALSE |
| 4 | ENSMUST00000178862.1 cds chromosome:GRCm38:6:41542163:41542176:1 gene:ENSMUSG00000094569.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:Trbd2 description:T cell receptor beta, D region 2 [Source:MGI Symbol;Acc:MGI:4439727] | 14 | 3AC456B17D30D58ED16896AE1FB699C7 | TRUE | 1 | FALSE |
| 5 | ENSMUST00000179520.1 cds chromosome:GRCm38:12:113430528:113430538:-1 gene:ENSMUSG00000094028.1 gene_biotype:IG_D_gene transcript_biotype:IG_D_gene gene_symbol:Ighd4-1 description:immunoglobulin heavy diversity 4-1 [Source:MGI Symbol;Acc:MGI:4439801] | 11 | 1B514044F866F7820E6F5A474D288132 | TRUE | 1 | FALSE |
| 6 | ENSMUST00000179883.1 cds chromosome:GRCm38:12:113448214:113448229:-1 gene:ENSMUSG00000094552.1 gene_biotype:IG_D_gene transcript_biotype:IG_D_gene gene_symbol:Ighd3-2 description:immunoglobulin heavy diversity 3-2 [Source:MGI Symbol;Acc:MGI:4439707] | 16 | 5A0A74B83FAC2445F1585E0D3E36018E | TRUE | 1 | FALSE |
nsequences
nrow(transcripts)58141
68381
# Assign a random TPM value to each non-duplicated transcript sequence
TPMs <- rep(0, nrow(transcripts))
TPMs[!transcripts$Duplicate] <- rexp(nsequences)
# those that are duplicated are given '0' TPM values. true.counts <- simReads(
# the transcript database and the wanted abundances
'/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus.GRCm38.cds.all.LINEWRAP.fa',
TPMs,
# the name of the output
"/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus_simRNA_1mReads",
# options on the output
library.size = 1000000,
read.length = 100,
truth.in.read.names = FALSE,
# simulating sequencing errors
simulate.sequencing.error = TRUE,
quality.reference = NULL,
# parameters for generating paired-end reads.
paired.end = TRUE,
fragment.length.min = 125,
fragment.length.max = 500,
fragment.length.mean = 250,
fragment.length.sd = 40,
# manipulating transcript names
simplify.transcript.names = FALSE)
write.table(x = true.counts, file = '/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus.GRCm38.1m_isoform_true_expression.txt', sep = '\t', quote = F, row.names = F)true.counts <- simReads(
# the transcript database and the wanted abundances
'/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus.GRCm38.cds.all.LINEWRAP.fa',
TPMs,
# the name of the output
"/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus_simRNA_30mReads",
# options on the output
library.size = 30000000,
read.length = 100,
truth.in.read.names = FALSE,
# simulating sequencing errors
simulate.sequencing.error = TRUE,
quality.reference = NULL,
# parameters for generating paired-end reads.
paired.end = TRUE,
fragment.length.min = 125,
fragment.length.max = 500,
fragment.length.mean = 250,
fragment.length.sd = 40,
# manipulating transcript names
simplify.transcript.names = FALSE)
write.table(x = true.counts, file = '/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus.GRCm38.30m_isoform_true_expression.txt', sep = '\t', quote = F, row.names = F)Files used:
wget http://ftp.ensembl.org/pub/release-104/fasta/homo_sapiens/cds/Homo_sapiens.GRCh38.cds.all.fa.gz
gunzip Homo_sapiens.GRCh38.cds.all.fa.gz
reformat.sh in=Homo_sapiens.GRCh38.cds.all.fa out=Homo_sapiens.GRCh38.cds.all.LINEWRAP.fa fastawrap=80
BBMap is the source of reformat.sh. This script takes a single line fasta and turns it to 80 character line wrapped fasta.
simReads can't use single line FASTA. An error is thrown.
# Scan through the fasta file to get transcript names and lengths
transcripts <- scanFasta('/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens.GRCh38.cds.all.LINEWRAP.fa')
nsequences <- nrow(transcripts) - sum(transcripts$Duplicate)
Warning: 36435 duplicate sequences were found in the input. Please check the summary table.
head(transcripts)| TranscriptID | Length | MD5 | Unique | Occurrence | Duplicated | |
|---|---|---|---|---|---|---|
| <chr> | <int> | <chr> | <lgl> | <int> | <lgl> | |
| 1 | ENST00000631435.1 cds chromosome:GRCh38:CHR_HSCHR7_2_CTG6:142847306:142847317:1 gene:ENSG00000282253.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRBD1 description:T cell receptor beta diversity 1 [Source:HGNC Symbol;Acc:HGNC:12158] | 12 | 1F47B55923E2D23090F894C439974B55 | FALSE | 2 | FALSE |
| 2 | ENST00000415118.1 cds chromosome:GRCh38:14:22438547:22438554:1 gene:ENSG00000223997.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRDD1 description:T cell receptor delta diversity 1 [Source:HGNC Symbol;Acc:HGNC:12254] | 8 | 940CA0BE6717D9330B746F88BBF1FC93 | TRUE | 1 | FALSE |
| 3 | ENST00000448914.1 cds chromosome:GRCh38:14:22449113:22449125:1 gene:ENSG00000228985.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRDD3 description:T cell receptor delta diversity 3 [Source:HGNC Symbol;Acc:HGNC:12256] | 13 | 8DFFD69B083D7B2ACC3879B5936B6D16 | TRUE | 1 | FALSE |
| 4 | ENST00000434970.2 cds chromosome:GRCh38:14:22439007:22439015:1 gene:ENSG00000237235.2 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRDD2 description:T cell receptor delta diversity 2 [Source:HGNC Symbol;Acc:HGNC:12255] | 9 | 00786AB95D4186F71651FE88A9AFD016 | TRUE | 1 | FALSE |
| 5 | ENST00000632684.1 cds chromosome:GRCh38:7:142786213:142786224:1 gene:ENSG00000282431.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRBD1 description:T cell receptor beta diversity 1 [Source:HGNC Symbol;Acc:HGNC:12158] | 12 | 1F47B55923E2D23090F894C439974B55 | FALSE | 2 | TRUE |
| 6 | ENST00000633010.1 cds chromosome:GRCh38:CHR_HSCHR14_3_CTG1:105895279:105895294:-1 gene:ENSG00000282274.1 gene_biotype:IG_D_gene transcript_biotype:IG_D_gene gene_symbol:IGHD4-17 description:immunoglobulin heavy diversity 4-17 [Source:HGNC Symbol;Acc:HGNC:5503] | 16 | 71E47CB9367D2425A5C45BEB905C00FB | FALSE | 2 | FALSE |
nsequences
nrow(transcripts)93450
116646
# Assign a random TPM value to each non-duplicated transcript sequence
TPMs <- rep(0, nrow(transcripts))
TPMs[!transcripts$Duplicate] <- rexp(nsequences)
# those that are duplicated are given '0' TPM values. true.counts <- simReads(
# the transcript database and the wanted abundances
'/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens.GRCh38.cds.all.LINEWRAP.fa',
TPMs,
# the name of the output
"/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens_simRNA_1mReads",
# options on the output
library.size = 1000000,
read.length = 100,
truth.in.read.names = FALSE,
# simulating sequencing errors
simulate.sequencing.error = TRUE,
quality.reference = NULL,
# parameters for generating paired-end reads.
paired.end = TRUE,
fragment.length.min = 125,
fragment.length.max = 500,
fragment.length.mean = 250,
fragment.length.sd = 40,
# manipulating transcript names
simplify.transcript.names = FALSE)
write.table(x = true.counts, file = '/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens.GRCh38.1m_isoform_true_expression.txt', sep = '\t', quote = F, row.names = F)true.counts <- simReads(
# the transcript database and the wanted abundances
'/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens.GRCh38.cds.all.LINEWRAP.fa',
TPMs,
# the name of the output
"/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens_simRNA_30mReads",
# options on the output
library.size = 30000000,
read.length = 100,
truth.in.read.names = FALSE,
# simulating sequencing errors
simulate.sequencing.error = TRUE,
quality.reference = NULL,
# parameters for generating paired-end reads.
paired.end = TRUE,
fragment.length.min = 125,
fragment.length.max = 500,
fragment.length.mean = 250,
fragment.length.sd = 40,
# manipulating transcript names
simplify.transcript.names = FALSE)
write.table(x = true.counts, file = '/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens.GRCh38.30m_isoform_true_expression.txt', sep = '\t', quote = F, row.names = F)mouse_simulated_truth_1m <- read.delim('/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus.GRCm38.1m_isoform_true_expression.txt', sep="\t", header=T)
mouse_simulated_test_1m <- read.delim('/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RESULTS/RNA_mouse_nxf_bench/Mus_musculus_simRNA_1mReads/Mus_musculus_simRNA_1mReads.isoforms.results', sep="\t", header=T)
mouse_simulated_truth_30m <- read.delim('/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RNA/mouse_cds/Mus_musculus.GRCm38.30m_isoform_true_expression.txt', sep="\t", header=T)
mouse_simulated_test_30m <- read.delim('/projects/omics_share/mouse/GRCm38/supporting_files/benchmarking_data/RESULTS/RNA_mouse_nxf_bench/Mus_musculus_simRNA_30mReads/Mus_musculus_simRNA_30mReads.isoforms.results', sep="\t", header=T)
head(mouse_simulated_truth_1m)
head(mouse_simulated_test_1m)| TranscriptID | Length | NReads | |
|---|---|---|---|
| <fct> | <int> | <int> | |
| 1 | ENSMUST00000177564.1 cds chromosome:GRCm38:14:54122226:54122241:1 gene:ENSMUSG00000096176.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:Trdd2 description:T cell receptor delta diversity 2 [Source:MGI Symbol;Acc:MGI:4439546] | 16 | 0 |
| 2 | ENSMUST00000196221.1 cds chromosome:GRCm38:14:54113468:54113476:1 gene:ENSMUSG00000096749.2 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:Trdd1 description:T cell receptor delta diversity 1 [Source:MGI Symbol;Acc:MGI:4439547] | 9 | 0 |
| 3 | ENSMUST00000178537.1 cds chromosome:GRCm38:6:41533201:41533212:1 gene:ENSMUSG00000095668.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:Trbd1 description:T cell receptor beta, D region 1 [Source:MGI Symbol;Acc:MGI:4439571] | 12 | 0 |
| 4 | ENSMUST00000178862.1 cds chromosome:GRCm38:6:41542163:41542176:1 gene:ENSMUSG00000094569.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:Trbd2 description:T cell receptor beta, D region 2 [Source:MGI Symbol;Acc:MGI:4439727] | 14 | 0 |
| 5 | ENSMUST00000179520.1 cds chromosome:GRCm38:12:113430528:113430538:-1 gene:ENSMUSG00000094028.1 gene_biotype:IG_D_gene transcript_biotype:IG_D_gene gene_symbol:Ighd4-1 description:immunoglobulin heavy diversity 4-1 [Source:MGI Symbol;Acc:MGI:4439801] | 11 | 0 |
| 6 | ENSMUST00000179883.1 cds chromosome:GRCm38:12:113448214:113448229:-1 gene:ENSMUSG00000094552.1 gene_biotype:IG_D_gene transcript_biotype:IG_D_gene gene_symbol:Ighd3-2 description:immunoglobulin heavy diversity 3-2 [Source:MGI Symbol;Acc:MGI:4439707] | 16 | 0 |
| transcript_id | gene_id | length | effective_length | expected_count | TPM | FPKM | IsoPct | |
|---|---|---|---|---|---|---|---|---|
| <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | |
| 1 | ENSMUST00000000001_Gnai3-201 | ENSMUSG00000000001_Gnai3 | 3262 | 3013.28 | 0 | 0.00 | 0.00 | 0 |
| 2 | ENSMUST00000000003_Pbsn-201 | ENSMUSG00000000003_Pbsn | 902 | 653.28 | 1 | 6.18 | 4.43 | 100 |
| 3 | ENSMUST00000114041_Pbsn-202 | ENSMUSG00000000003_Pbsn | 697 | 448.30 | 0 | 0.00 | 0.00 | 0 |
| 4 | ENSMUST00000000028_Cdc45-201 | ENSMUSG00000000028_Cdc45 | 2143 | 1894.28 | 0 | 0.00 | 0.00 | 0 |
| 5 | ENSMUST00000096990_Cdc45-202 | ENSMUSG00000000028_Cdc45 | 1747 | 1498.28 | 7 | 18.87 | 13.51 | 100 |
| 6 | ENSMUST00000115585_Cdc45-203 | ENSMUSG00000000028_Cdc45 | 832 | 583.29 | 0 | 0.00 | 0.00 | 0 |
mouse_simulated_truth_1m_clean <- mouse_simulated_truth_1m %>%
separate( TranscriptID, into=c("transcript_id", "CDS", 'location', 'gene_id', 'gene_biotype', 'transcript_biotype', 'gene_symbol'), sep= " ", extra = 'drop') %>%
mutate(location = str_remove(location, "chromosome:GRCm38:"),
gene_id = str_remove(gene_id, 'gene:'),
gene_biotype = str_remove(gene_biotype, 'gene_biotype:'),
transcript_biotype = str_remove(transcript_biotype, 'transcript_biotype:'),
gene_symbol = str_remove(gene_symbol, 'gene_symbol:'),
transcript_id = sub('\\.[0-9]*$', '', transcript_id),
gene_id = sub('\\.[0-9]*$', '', gene_id))mouse_simulated_truth_30m_clean <- mouse_simulated_truth_30m %>%
separate( TranscriptID, into=c("transcript_id", "CDS", 'location', 'gene_id', 'gene_biotype', 'transcript_biotype', 'gene_symbol'), sep= " ", extra = 'drop') %>%
mutate(location = str_remove(location, "chromosome:GRCm38:"),
gene_id = str_remove(gene_id, 'gene:'),
gene_biotype = str_remove(gene_biotype, 'gene_biotype:'),
transcript_biotype = str_remove(transcript_biotype, 'transcript_biotype:'),
gene_symbol = str_remove(gene_symbol, 'gene_symbol:'),
transcript_id = sub('\\.[0-9]*$', '', transcript_id),
gene_id = sub('\\.[0-9]*$', '', gene_id))
#This was used for ensembl CDS file. That file included additional transcripts relative to what we run against in the pipeline. library(stringi)
mouse_simulated_test_1m <- cbind(mouse_simulated_test_1m, `colnames<-`(stri_split_fixed(mouse_simulated_test_1m$transcript_id, '_', n = 2, simplify = TRUE), c("TranscriptID", "gene_symbol")))
mouse_simulated_test_30m <- cbind(mouse_simulated_test_30m, `colnames<-`(stri_split_fixed(mouse_simulated_test_30m$transcript_id, '_', n = 2, simplify = TRUE), c("TranscriptID", "gene_symbol")))
head(mouse_simulated_test_1m)| transcript_id | gene_id | length | effective_length | expected_count | TPM | FPKM | IsoPct | TranscriptID | gene_symbol | |
|---|---|---|---|---|---|---|---|---|---|---|
| <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> | <fct> | |
| 1 | ENSMUST00000000001_Gnai3-201 | ENSMUSG00000000001_Gnai3 | 3262 | 3013.28 | 0 | 0.00 | 0.00 | 0 | ENSMUST00000000001 | Gnai3-201 |
| 2 | ENSMUST00000000003_Pbsn-201 | ENSMUSG00000000003_Pbsn | 902 | 653.28 | 1 | 6.18 | 4.43 | 100 | ENSMUST00000000003 | Pbsn-201 |
| 3 | ENSMUST00000114041_Pbsn-202 | ENSMUSG00000000003_Pbsn | 697 | 448.30 | 0 | 0.00 | 0.00 | 0 | ENSMUST00000114041 | Pbsn-202 |
| 4 | ENSMUST00000000028_Cdc45-201 | ENSMUSG00000000028_Cdc45 | 2143 | 1894.28 | 0 | 0.00 | 0.00 | 0 | ENSMUST00000000028 | Cdc45-201 |
| 5 | ENSMUST00000096990_Cdc45-202 | ENSMUSG00000000028_Cdc45 | 1747 | 1498.28 | 7 | 18.87 | 13.51 | 100 | ENSMUST00000096990 | Cdc45-202 |
| 6 | ENSMUST00000115585_Cdc45-203 | ENSMUSG00000000028_Cdc45 | 832 | 583.29 | 0 | 0.00 | 0.00 | 0 | ENSMUST00000115585 | Cdc45-203 |
mouse_1m_joined_data <- dplyr::left_join(mouse_simulated_truth_1m_clean, mouse_simulated_test_1m, by = c('transcript_id' = 'TranscriptID'))
mouse_30m_joined_data <- dplyr::left_join(mouse_simulated_truth_30m_clean, mouse_simulated_test_30m, by = c('transcript_id' = 'TranscriptID'))head(mouse_1m_joined_data)| transcript_id | CDS | location | gene_id.x | gene_biotype | transcript_biotype | gene_symbol.x | Length | NReads | transcript_id.y | gene_id.y | length | effective_length | expected_count | TPM | FPKM | IsoPct | gene_symbol.y | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <int> | <int> | <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> | |
| 1 | ENSMUST00000177564 | cds | 14:54122226:54122241:1 | ENSMUSG00000096176 | TR_D_gene | TR_D_gene | Trdd2 | 16 | 0 | ENSMUST00000177564_Trdd2-201 | ENSMUSG00000096176_Trdd2 | 16 | 0 | 0 | 0 | 0 | 0 | Trdd2-201 |
| 2 | ENSMUST00000196221 | cds | 14:54113468:54113476:1 | ENSMUSG00000096749 | TR_D_gene | TR_D_gene | Trdd1 | 9 | 0 | ENSMUST00000196221_Trdd1-202 | ENSMUSG00000096749_Trdd1 | 9 | 0 | 0 | 0 | 0 | 0 | Trdd1-202 |
| 3 | ENSMUST00000178537 | cds | 6:41533201:41533212:1 | ENSMUSG00000095668 | TR_D_gene | TR_D_gene | Trbd1 | 12 | 0 | ENSMUST00000178537_Trbd1-201 | ENSMUSG00000095668_Trbd1 | 12 | 0 | 0 | 0 | 0 | 0 | Trbd1-201 |
| 4 | ENSMUST00000178862 | cds | 6:41542163:41542176:1 | ENSMUSG00000094569 | TR_D_gene | TR_D_gene | Trbd2 | 14 | 0 | ENSMUST00000178862_Trbd2-201 | ENSMUSG00000094569_Trbd2 | 14 | 0 | 0 | 0 | 0 | 0 | Trbd2-201 |
| 5 | ENSMUST00000179520 | cds | 12:113430528:113430538:-1 | ENSMUSG00000094028 | IG_D_gene | IG_D_gene | Ighd4-1 | 11 | 0 | ENSMUST00000179520_Ighd4-1-201 | ENSMUSG00000094028_Ighd4-1 | 11 | 0 | 0 | 0 | 0 | 0 | Ighd4-1-201 |
| 6 | ENSMUST00000179883 | cds | 12:113448214:113448229:-1 | ENSMUSG00000094552 | IG_D_gene | IG_D_gene | Ighd3-2 | 16 | 0 | ENSMUST00000179883_Ighd3-2-201 | ENSMUSG00000094552_Ighd3-2 | 16 | 0 | 0 | 0 | 0 | 0 | Ighd3-2-201 |
head(mouse_1m_joined_data)| transcript_id | CDS | location | gene_id.x | gene_biotype | transcript_biotype | gene_symbol.x | Length | NReads | transcript_id.y | gene_id.y | length | effective_length | expected_count | TPM | FPKM | IsoPct | gene_symbol.y | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <int> | <int> | <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> | |
| 1 | ENSMUST00000177564 | cds | 14:54122226:54122241:1 | ENSMUSG00000096176 | TR_D_gene | TR_D_gene | Trdd2 | 16 | 0 | ENSMUST00000177564_Trdd2-201 | ENSMUSG00000096176_Trdd2 | 16 | 0 | 0 | 0 | 0 | 0 | Trdd2-201 |
| 2 | ENSMUST00000196221 | cds | 14:54113468:54113476:1 | ENSMUSG00000096749 | TR_D_gene | TR_D_gene | Trdd1 | 9 | 0 | ENSMUST00000196221_Trdd1-202 | ENSMUSG00000096749_Trdd1 | 9 | 0 | 0 | 0 | 0 | 0 | Trdd1-202 |
| 3 | ENSMUST00000178537 | cds | 6:41533201:41533212:1 | ENSMUSG00000095668 | TR_D_gene | TR_D_gene | Trbd1 | 12 | 0 | ENSMUST00000178537_Trbd1-201 | ENSMUSG00000095668_Trbd1 | 12 | 0 | 0 | 0 | 0 | 0 | Trbd1-201 |
| 4 | ENSMUST00000178862 | cds | 6:41542163:41542176:1 | ENSMUSG00000094569 | TR_D_gene | TR_D_gene | Trbd2 | 14 | 0 | ENSMUST00000178862_Trbd2-201 | ENSMUSG00000094569_Trbd2 | 14 | 0 | 0 | 0 | 0 | 0 | Trbd2-201 |
| 5 | ENSMUST00000179520 | cds | 12:113430528:113430538:-1 | ENSMUSG00000094028 | IG_D_gene | IG_D_gene | Ighd4-1 | 11 | 0 | ENSMUST00000179520_Ighd4-1-201 | ENSMUSG00000094028_Ighd4-1 | 11 | 0 | 0 | 0 | 0 | 0 | Ighd4-1-201 |
| 6 | ENSMUST00000179883 | cds | 12:113448214:113448229:-1 | ENSMUSG00000094552 | IG_D_gene | IG_D_gene | Ighd3-2 | 16 | 0 | ENSMUST00000179883_Ighd3-2-201 | ENSMUSG00000094552_Ighd3-2 | 16 | 0 | 0 | 0 | 0 | 0 | Ighd3-2-201 |
plot(mouse_1m_joined_data$NReads, mouse_1m_joined_data$expected_count, xlab = 'True Read Counts', ylab = 'RSEM Expected Count')
plot(mouse_30m_joined_data$NReads, mouse_30m_joined_data$expected_count, xlab = 'True Read Counts', ylab = 'RSEM Expected Count')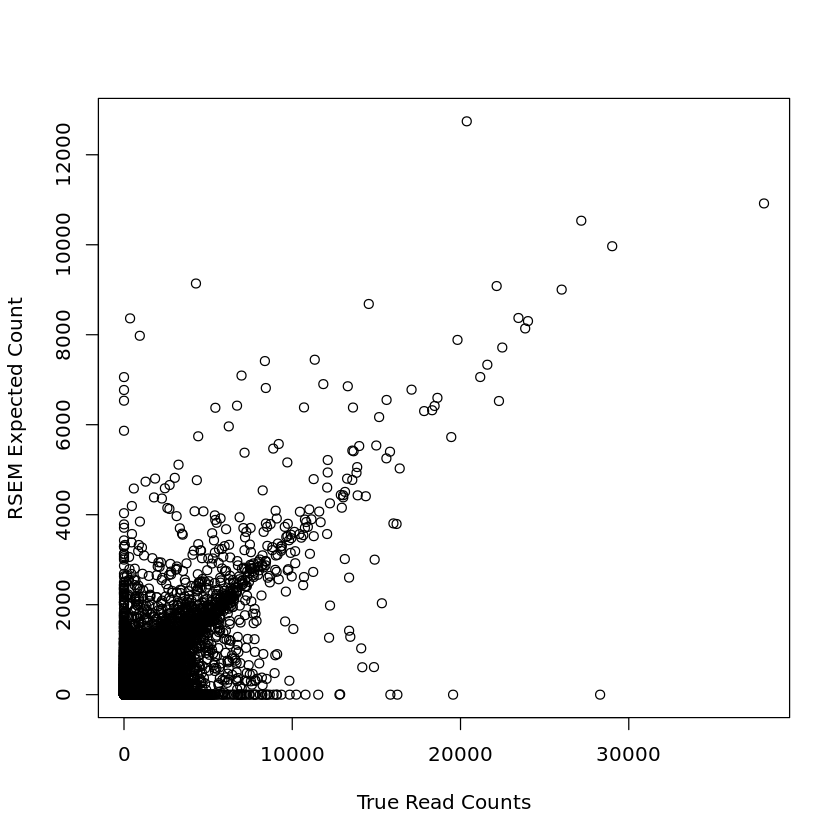
mouse_1m_joined_data %>% filter(is.na(effective_length))
mouse_30m_joined_data %>% filter(is.na(effective_length))
# checking the join. | transcript_id | CDS | location | gene_id.x | gene_biotype | transcript_biotype | gene_symbol.x | Length | NReads | transcript_id.y | gene_id.y | length | effective_length | expected_count | TPM | FPKM | IsoPct | gene_symbol.y |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <int> | <int> | <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> |
| transcript_id | CDS | location | gene_id.x | gene_biotype | transcript_biotype | gene_symbol.x | Length | NReads | transcript_id.y | gene_id.y | length | effective_length | expected_count | TPM | FPKM | IsoPct | gene_symbol.y |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <int> | <int> | <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> |
I have an Ensembl ID, what can I tell about it from the ID?
An Ensembl stable ID consists of five parts: ENS(species)(object type)(identifier).(version).
The first part, 'ENS', tells you that it's an Ensembl ID
The second part is a three-letter species code. For human, there is no species code so IDs are in the form ENS(object type)(identifier).(version). A list of the other species codes can be found here.
The third part is a one- or two-letter object type. For example E for exon, FM for protein family, G for gene, GT for gene tree, P for protein, R for regulatory feature and T for transcript.
The identifier is the number to that object. Combinations of prefixes and identifiers are unique.
Versions indicate how many times that model has changed during its time in Ensembl. This document explains how we determine that a model has changed sufficiently to update version number. History pages for features show you when these changes took place.
Using this information we can make assertions about an Ensembl ID. For example ENSMUSG00000017167.6. From this we can see that it's an Ensembl ID (ENS), from mouse (MUS), it's a gene (G) and it's on its sixth version (.6).
human_simulated_truth_1m <- read.delim('/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens.GRCh38.1m_isoform_true_expression.txt', sep="\t", header=T)
human_simulated_test_1m <- read.delim('/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RESULTS/RNA_human_nxf_bench/Homo_sapiens_simRNA_1mReads/Homo_sapiens_simRNA_1mReads.isoforms.results', sep="\t", header=T)
human_simulated_truth_30m <- read.delim('/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RNA/human_cds/Homo_sapiens.GRCh38.30m_isoform_true_expression.txt', sep="\t", header=T)
human_simulated_test_30m <- read.delim('/projects/omics_share/human/GRCh38/supporting_files/benchmarking_data/RESULTS/RNA_human_nxf_bench/Homo_sapiens_simRNA_30mReads/Homo_sapiens_simRNA_30mReads.isoforms.results', sep="\t", header=T)
head(human_simulated_test_1m)
head(human_simulated_truth_1m)| transcript_id | gene_id | length | effective_length | expected_count | TPM | FPKM | IsoPct | |
|---|---|---|---|---|---|---|---|---|
| <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | |
| 1 | ENST00000373020_TSPAN6-201 | ENSG00000000003_TSPAN6 | 3768 | 3519.41 | 1.23 | 1.33 | 1.01 | 2.85 |
| 2 | ENST00000494424_TSPAN6-202 | ENSG00000000003_TSPAN6 | 820 | 571.41 | 0.00 | 0.00 | 0.00 | 0.00 |
| 3 | ENST00000496771_TSPAN6-203 | ENSG00000000003_TSPAN6 | 1025 | 776.41 | 0.00 | 0.00 | 0.00 | 0.00 |
| 4 | ENST00000612152_TSPAN6-204 | ENSG00000000003_TSPAN6 | 3796 | 3547.41 | 0.00 | 0.00 | 0.00 | 0.00 |
| 5 | ENST00000614008_TSPAN6-205 | ENSG00000000003_TSPAN6 | 900 | 651.41 | 7.77 | 45.42 | 34.38 | 97.15 |
| 6 | ENST00000373031_TNMD-201 | ENSG00000000005_TNMD | 1205 | 956.41 | 0.00 | 0.00 | 0.00 | 0.00 |
| TranscriptID | Length | NReads | |
|---|---|---|---|
| <fct> | <int> | <int> | |
| 1 | ENST00000631435.1 cds chromosome:GRCh38:CHR_HSCHR7_2_CTG6:142847306:142847317:1 gene:ENSG00000282253.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRBD1 description:T cell receptor beta diversity 1 [Source:HGNC Symbol;Acc:HGNC:12158] | 12 | 0 |
| 2 | ENST00000415118.1 cds chromosome:GRCh38:14:22438547:22438554:1 gene:ENSG00000223997.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRDD1 description:T cell receptor delta diversity 1 [Source:HGNC Symbol;Acc:HGNC:12254] | 8 | 0 |
| 3 | ENST00000448914.1 cds chromosome:GRCh38:14:22449113:22449125:1 gene:ENSG00000228985.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRDD3 description:T cell receptor delta diversity 3 [Source:HGNC Symbol;Acc:HGNC:12256] | 13 | 0 |
| 4 | ENST00000434970.2 cds chromosome:GRCh38:14:22439007:22439015:1 gene:ENSG00000237235.2 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRDD2 description:T cell receptor delta diversity 2 [Source:HGNC Symbol;Acc:HGNC:12255] | 9 | 0 |
| 5 | ENST00000632684.1 cds chromosome:GRCh38:7:142786213:142786224:1 gene:ENSG00000282431.1 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene gene_symbol:TRBD1 description:T cell receptor beta diversity 1 [Source:HGNC Symbol;Acc:HGNC:12158] | 12 | 0 |
| 6 | ENST00000633010.1 cds chromosome:GRCh38:CHR_HSCHR14_3_CTG1:105895279:105895294:-1 gene:ENSG00000282274.1 gene_biotype:IG_D_gene transcript_biotype:IG_D_gene gene_symbol:IGHD4-17 description:immunoglobulin heavy diversity 4-17 [Source:HGNC Symbol;Acc:HGNC:5503] | 16 | 0 |
human_simulated_truth_1m_clean <- human_simulated_truth_1m %>%
separate( TranscriptID, into=c("transcript_id", "CDS", 'location', 'gene_id', 'gene_biotype', 'transcript_biotype', 'gene_symbol'), sep= " ", extra = 'drop') %>%
mutate(location = str_remove(location, "chromosome:GRCh38:"),
gene_id = str_remove(gene_id, 'gene:'),
gene_biotype = str_remove(gene_biotype, 'gene_biotype:'),
transcript_biotype = str_remove(transcript_biotype, 'transcript_biotype:'),
gene_symbol = str_remove(gene_symbol, 'gene_symbol:'),
transcript_id = sub('\\.[0-9]*$', '', transcript_id),
gene_id = sub('\\.[0-9]*$', '', gene_id))
#This was used for ensembl CDS file. That file included additional transcripts relative to what we run against in the pipeline. Warning message:
“Expected 7 pieces. Missing pieces filled with `NA` in 387 rows [776, 777, 778, 779, 780, 961, 1182, 1183, 1205, 1206, 1248, 1256, 1364, 1365, 1444, 1445, 1523, 1524, 1630, 1720, ...].”
human_simulated_truth_30m_clean <- human_simulated_truth_30m %>%
separate( TranscriptID, into=c("transcript_id", "CDS", 'location', 'gene_id', 'gene_biotype', 'transcript_biotype', 'gene_symbol'), sep= " ", extra = 'drop') %>%
mutate(location = str_remove(location, "chromosome:GRCh38:"),
gene_id = str_remove(gene_id, 'gene:'),
gene_biotype = str_remove(gene_biotype, 'gene_biotype:'),
transcript_biotype = str_remove(transcript_biotype, 'transcript_biotype:'),
gene_symbol = str_remove(gene_symbol, 'gene_symbol:'),
transcript_id = sub('\\.[0-9]*$', '', transcript_id),
gene_id = sub('\\.[0-9]*$', '', gene_id))
#This was used for ensembl CDS file. That file included additional transcripts relative to what we run against in the pipeline. Warning message:
“Expected 7 pieces. Missing pieces filled with `NA` in 387 rows [776, 777, 778, 779, 780, 961, 1182, 1183, 1205, 1206, 1248, 1256, 1364, 1365, 1444, 1445, 1523, 1524, 1630, 1720, ...].”
head(human_simulated_truth_1m_clean)| transcript_id | CDS | location | gene_id | gene_biotype | transcript_biotype | gene_symbol | Length | NReads | |
|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <int> | <int> | |
| 1 | ENST00000631435 | cds | CHR_HSCHR7_2_CTG6:142847306:142847317:1 | ENSG00000282253 | TR_D_gene | TR_D_gene | TRBD1 | 12 | 0 |
| 2 | ENST00000415118 | cds | 14:22438547:22438554:1 | ENSG00000223997 | TR_D_gene | TR_D_gene | TRDD1 | 8 | 0 |
| 3 | ENST00000448914 | cds | 14:22449113:22449125:1 | ENSG00000228985 | TR_D_gene | TR_D_gene | TRDD3 | 13 | 0 |
| 4 | ENST00000434970 | cds | 14:22439007:22439015:1 | ENSG00000237235 | TR_D_gene | TR_D_gene | TRDD2 | 9 | 0 |
| 5 | ENST00000632684 | cds | 7:142786213:142786224:1 | ENSG00000282431 | TR_D_gene | TR_D_gene | TRBD1 | 12 | 0 |
| 6 | ENST00000633010 | cds | CHR_HSCHR14_3_CTG1:105895279:105895294:-1 | ENSG00000282274 | IG_D_gene | IG_D_gene | IGHD4-17 | 16 | 0 |
library(stringi)
human_simulated_test_1m <- cbind(human_simulated_test_1m, `colnames<-`(stri_split_fixed(human_simulated_test_1m$transcript_id, '_', n = 2, simplify = TRUE), c("TranscriptID", "gene_symbol")))
human_simulated_test_30m <- cbind(human_simulated_test_30m, `colnames<-`(stri_split_fixed(human_simulated_test_30m$transcript_id, '_', n = 2, simplify = TRUE), c("TranscriptID", "gene_symbol")))
human_1m_joined_data <- dplyr::left_join(human_simulated_truth_1m_clean, human_simulated_test_1m, by = c('transcript_id' = 'TranscriptID'))
human_30m_joined_data <- dplyr::left_join(human_simulated_truth_30m_clean, human_simulated_test_30m, by = c('transcript_id' = 'TranscriptID'))head(human_1m_joined_data)| transcript_id | CDS | location | gene_id.x | gene_biotype | transcript_biotype | gene_symbol.x | Length | NReads | transcript_id.y | gene_id.y | length | effective_length | expected_count | TPM | FPKM | IsoPct | gene_symbol.y | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <int> | <int> | <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> | |
| 1 | ENST00000631435 | cds | CHR_HSCHR7_2_CTG6:142847306:142847317:1 | ENSG00000282253 | TR_D_gene | TR_D_gene | TRBD1 | 12 | 0 | ENST00000631435_TRBD1-201 | ENSG00000282253_TRBD1 | 12 | 0 | 0 | 0 | 0 | 0 | TRBD1-201 |
| 2 | ENST00000415118 | cds | 14:22438547:22438554:1 | ENSG00000223997 | TR_D_gene | TR_D_gene | TRDD1 | 8 | 0 | ENST00000415118_TRDD1-201 | ENSG00000223997_TRDD1 | 8 | 0 | 0 | 0 | 0 | 0 | TRDD1-201 |
| 3 | ENST00000448914 | cds | 14:22449113:22449125:1 | ENSG00000228985 | TR_D_gene | TR_D_gene | TRDD3 | 13 | 0 | ENST00000448914_TRDD3-201 | ENSG00000228985_TRDD3 | 13 | 0 | 0 | 0 | 0 | 0 | TRDD3-201 |
| 4 | ENST00000434970 | cds | 14:22439007:22439015:1 | ENSG00000237235 | TR_D_gene | TR_D_gene | TRDD2 | 9 | 0 | ENST00000434970_TRDD2-201 | ENSG00000237235_TRDD2 | 9 | 0 | 0 | 0 | 0 | 0 | TRDD2-201 |
| 5 | ENST00000632684 | cds | 7:142786213:142786224:1 | ENSG00000282431 | TR_D_gene | TR_D_gene | TRBD1 | 12 | 0 | ENST00000632684_TRBD1-202 | ENSG00000282431_TRBD1 | 12 | 0 | 0 | 0 | 0 | 0 | TRBD1-202 |
| 6 | ENST00000633010 | cds | CHR_HSCHR14_3_CTG1:105895279:105895294:-1 | ENSG00000282274 | IG_D_gene | IG_D_gene | IGHD4-17 | 16 | 0 | ENST00000633010_IGHD4-17-202 | ENSG00000282274_IGHD4-17 | 16 | 0 | 0 | 0 | 0 | 0 | IGHD4-17-202 |
plot(human_1m_joined_data$NReads, human_1m_joined_data$expected_count, xlab = 'True Read Counts', ylab = 'RSEM Expected Count')
plot(human_30m_joined_data$NReads, human_30m_joined_data$expected_count, xlab = 'True Read Counts', ylab = 'RSEM Expected Count')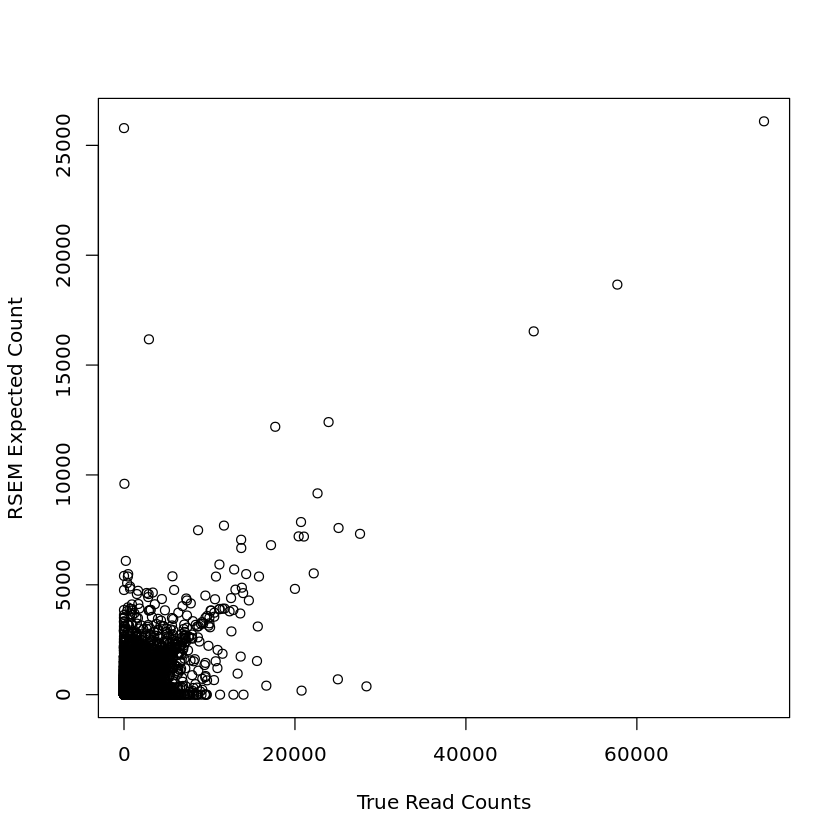
human_1m_joined_data %>% filter(is.na(effective_length))
human_30m_joined_data %>% filter(is.na(effective_length))
# checking the join. | transcript_id | CDS | location | gene_id.x | gene_biotype | transcript_biotype | gene_symbol.x | Length | NReads | transcript_id.y | gene_id.y | length | effective_length | expected_count | TPM | FPKM | IsoPct | gene_symbol.y |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <int> | <int> | <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> |
| transcript_id | CDS | location | gene_id.x | gene_biotype | transcript_biotype | gene_symbol.x | Length | NReads | transcript_id.y | gene_id.y | length | effective_length | expected_count | TPM | FPKM | IsoPct | gene_symbol.y |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <chr> | <int> | <int> | <fct> | <fct> | <int> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> |